Using a type of genetic screen that had previously been impossible in the mammalian brain, MIT neuroscientists have identified hundreds of genes that are necessary for neuron survival. They also used the same approach to identify genes that protect against the toxic effects of a mutant protein that causes Huntington’s disease.
These efforts yielded at least one promising drug target for Huntington’s: a family of genes that may normally help cells to break down the mutated huntingtin protein before it can aggregate and form the clumps seen in the brains of Huntington’s patients.
“These genes had never been linked to Huntington’s disease processes before. When we saw them, that was very exciting because we found not only one gene, but actually several of the same family, and also we saw them have an effect across two models of Huntington’s disease,” says Myriam Heiman, an associate professor of neuroscience in the Department of Brain and Cognitive Sciences and the senior author of the study.
The researchers’ new screening technique, which allowed them to assess all of the roughly 22,000 genes found in the mouse brain, could also be applied to other neurological disorders, including Alzheimer’s and Parkinson’s diseases, says Heiman, who is also a member of MIT’s Picower Institute for Learning and Memory and the Broad Institute of MIT and Harvard.
Broad Institute postdoc Mary Wertz is the lead author of the paper, which appears today in Neuron.
Genome-wide screen
For many decades, biologists have been performing screens in which they systematically knock out individual genes in model organisms such as mice, fruit flies, and the worm C. elegans, then observe the effects on cell survival. However, such screens have never been done in the mouse brain. One major reason for this is that delivering the molecular machinery required for these genetic manipulations is more difficult in the brain than elsewhere in the body.
“These unbiased genetic screens are very powerful, but the technical difficulty of doing it in the central nervous system at a genome-wide scale has never been overcome,” Heiman says.
In recent years, researchers at the Broad Institute have developed libraries of genetic material that can be used to turn off the expression of every gene found in the mouse genome. One of these libraries is based on short hairpin RNA (shRNA), which interferes with the messenger RNA that carries a particular gene’s information. Another makes use of CRISPR, a technique that can disrupt or delete specific genes in a cell. These libraries are delivered by viruses, each of which carry one element that targets a single gene.
The libraries were designed so that each of the approximately 22,000 mouse genes is targeted by four or five shRNAs or CRISPR components, so 80,000 to 100,000 viruses need to make it into the brain to ensure that all genes are hit at least once. The MIT team came up with a way to make their solution of viruses highly concentrated, and to inject them directly into the striatum of the brain. Using this approach, they were able to deliver one of the shRNA or CRISPR elements to about 25 percent of all of the cells in the striatum.
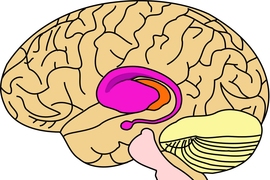
The researchers focused on the striatum, which is involved in regulating motor control, cognition, and emotion, because it is the brain region most affected by Huntington’s disease. It is also involved in Parkinson’s disease, as well as autism and drug addiction.
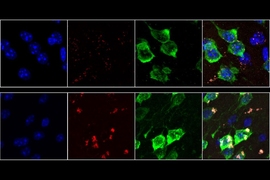
About seven months after the injection, the researchers sequenced all of the genomic DNA in the targeted striatal neurons. Their approach is based on the idea that if particular genes are necessary for neurons’ survival, any cell with those genes knocked out will die. Then, those shRNAs or CRISPR elements will be found at lower rates in the total population of cells.
The study turned up many genes that are necessary for any cell to survive, such as enzymes involved in cell metabolism or copying DNA into RNA. The findings also revealed genes that had been identified in previous studies of fruit flies and worms as being important for neuron function, such as genes involved the function of synapses (structures that allow neurons to communicate with each other).
However, a novel finding of this study was the identification of genes that hadn’t been linked to neuron survival before, Heiman says. Many of those were genes that code for metabolic proteins that are essential in cells that burn a lot of energy.
“What we interpret this to mean is that neurons in the mammalian brain are much more metabolically active and have a much higher dependency on these processes than for example, a neuron in C. elegans,” Heiman says.
William Yang, a professor of psychiatry and biobehavioral sciences at the University of California at Los Angeles, calls the new screening technique “a giant leap forward” for the field of brain research.
“Prior to this, people really could study the molecular function of genes gene-by-gene, or maybe a few genes at a time. This is a groundbreaking study because it demonstrates that you can perform genome-wide genetic screening in the mammalian central nervous system,” says Yang, who was not involved in the study.
Promising targets
The researchers then performed the same type of screen on two different mouse models of Huntington’s disease. These mouse models express the mutated form of the huntingtin protein, which forms clumps in the brains of Huntington’s patients. In this case, the researchers compared the results from the screen of the Huntington’s mice to normal mice. If any of the shRNA or CRISPR elements were found less frequently in the Huntington’s mice, that would suggest that those elements targeted genes that are helping to make cells more resistant to the toxic effects of the huntingtin protein, Heiman says.
One promising drug target that emerged from this screen is the Nme gene family, which has previously been linked to cancer metastasis, but not Huntington’s disease. The MIT team found that one of these genes, Nme1, regulates the expression of other genes that are involved in the proper disposal of proteins. The researchers hypothesize that without Nme1, these genes don’t get turned on as highly, allowing huntingtin to accumulate in the brain. They also showed that when Nme1 is overexpressed in the mouse models of Huntington’s, the Huntington’s symptoms appear to improve.
Although this gene hasn’t been linked to Huntington’s before, there have already been some efforts to develop compounds that target it, for use in treating cancer, Heiman says.
“This is very exciting to us because it’s theoretically a druggable compound,” she says. “If we can increase its activity with a small molecule, perhaps we can replicate the effect of genetic overexpression.”
The research was funded by the National Institutes of Health/National Institute of Neurological Disorders and Stroke, the JPB Foundation, the Bev Hartig Huntington’s Disease Foundation, a Fay/Frank Seed Award from the Brain Research Foundation, the Jeptha H. and Emily V. Wade Award, and the Hereditary Disease Foundation.