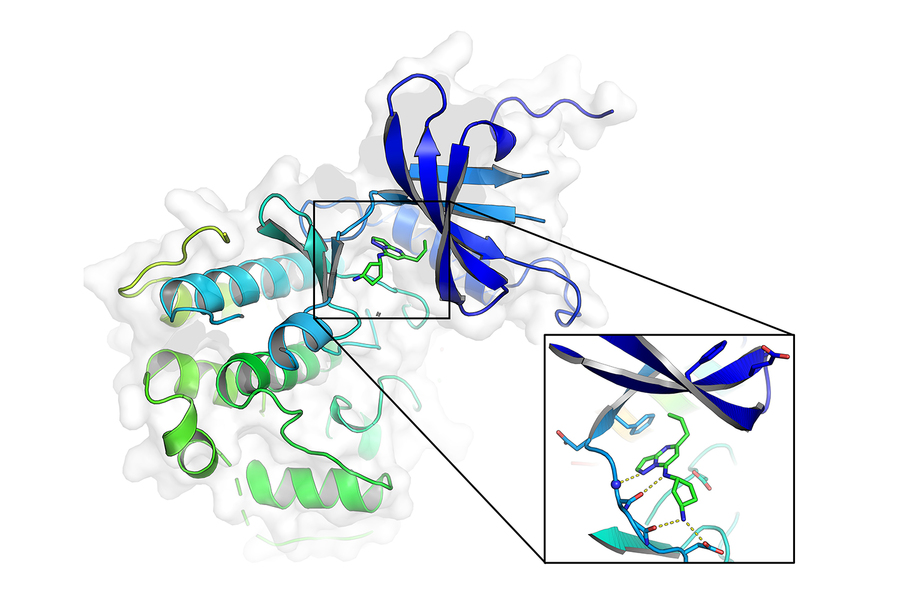
Prostate cancer growth is often driven by male sex hormones called androgens. Hormone therapy is commonly administered to lower the level of androgens in the body, but relapse is common when the cancer cells develop resistance to these therapies.
A multidisciplinary team of cancer researchers led by Angela Koehler, the Samuel A. Goldblith Career Development Professor in Applied Biology and a member of MIT’s Koch Institute for Integrative Cancer Research, has identified a small molecule that can selectively target a key protein involved in the stabilization of androgen receptor molecules.
Patients with advanced forms of prostate cancer are most commonly treated with androgen deprivation therapy, which lowers the level of male hormones such as testosterone through surgery or medication. However, patients invariably relapse from these therapies and develop castration-resistant prostate cancer (CRPC).
The next line of defense relies on drugs such as abiraterone or enzalutamide, which block testosterone and related androgens from binding to androgen receptors. Androgen receptors are proteins within cells that bind testosterone and ultimately upregulate the expression of genes that play a central role in prostate cancer development. Approximately half of CRPC patients respond to second-line drugs, although most relapse within one to two years.
Through a variety of means, cancer cells can develop other mechanisms to activate the androgen receptor pathway, frequently by expressing variant forms of androgen receptors that function even in the absence of androgens. There is no known small molecule drug that can inhibit androgen receptor variants, leaving few effective therapeutic options for CRPC patients.
In a study appearing in Cell Chemical Biology, the team identified a highly selective inhibitor of CDK9, which was not only highly potent against CRPC in cell lines and mouse models, but may have broad applicability to variety of cancers.
“Resistance to treatment is a common challenge in cancer care,” says postdoc Shelby Doyle PhD '20, who co-led the study as a part of her graduate thesis work in biological engineering. “Our compound could help combat that resistance and help us understand whether there is a more durable approach to chemotherapy in prostate cancer.”
Binding results
Drugs that block androgen receptor activity work by targeting the ligand binding domain. Analogous to a lock and key, the ligand binding domain provides a well-defined structure into which the small molecule drug can fit. But androgen receptor variants are missing the ligand binding domain, rendering androgen receptor-blocking drugs ineffective.
These “undruggable” androgen receptor variants are difficult to study with conventional methods. Traditional binding assays, which test small molecules against a purified protein of interest, depend on the presence of well-defined protein structures like a ligand binding domain to work. But when purified, androgen receptor variants lack such structure. Moreover, the purification process strips target proteins of associated cofactors, molecules that interact closely with the target protein and thus could serve as indirect targets.
The Koehler lab specializes in small-molecule microarray (SMM) platforms that screen libraries of small molecules against proteins together with their suites of cofactors. The cofactors themselves not only present additional targets, but their presence means that the androgen receptor variant molecule is more likely to adopt a defined structure, similar to that which it takes in the cell. Using the SMM platform to screen a library of 50,000 compounds for interactions with androgen receptor variants, researchers were able to identify a handful of binders. One of them, KI-ARv-03, proved to be very potent against CRPC cell lines.
“Our lead compound, KI-ARv-03, was particularly exciting to us because it was able to reduce androgen receptor activity by reducing levels of both androgen receptor and the chemoresistance-associated variant of androgen receptor in CPRC models,” says Doyle.
Experiments revealed that KI-ARv-03 does not target the androgen receptor protein directly, but binds to one of its cofactors, cyclin-dependent kinase 9 (CDK9). CDK9 is one of a family of proteins that regulate a variety of important cell processes, such as the cell cycle or gene transcription, and plays a role in stabilizing the androgen receptor molecule. By inhibiting CDK9, KI-ARv-03 destabilizes androgen receptor proteins and curbs the expression of oncogenes.
A recipe for optimization
CDK9 inhibitors have shown promise as potential cancer therapies, since they exploit a vulnerability unique to cancer cells, known as oncogene addiction. Cancer cells can overcome disordered or dysfunctional pathways by significantly increasing the transcription of certain genes that allow them to survive and grow. However, oncogene addiction leaves cancer cells vulnerable in a way healthy cells are not: If these oncogenic pathways are inhibited even for a short period of time, the cancer cells die.
Despite this promise, CDK9 inhibitors have had limited clinical success, likely in part because they bind to multiple members of the CDK family and result in high levels of toxicity. However, the researchers found that KI-ARv-03 is selective for CDK9 to an unusually high degree, and is unlikely to interact with other members of the CDK family.
“Kinase inhibitors often target multiple kinases because of the high structural similarity of these proteins, and it is very challenging to design selective binders for these,” explains research scientist André Richters, a chemist in the Koehler lab who co-led the study with Doyle. “Multiple rounds of chemical optimization and retesting are usually needed to improve selectivity while retaining potency. Success, however, is not guaranteed. The discovery of an ultra-selective and simultaneously potent kinase inhibitor such as KI-ARv-03 as a direct result of screening is extremely rare.”
Kronos Bio, an MIT spinout co-founded by Koehler, developed a more powerful version of the CDK9 inhibitor, KB-0742. Using proprietary technology, the company has optimized the KI-ARv-03 compound to make it more potent, while retaining the original selectivity uncovered by the MIT team. Preclinical tests in cell lines and mouse models reveal significantly reduced tumor growth in CPRC models and other oncogene-addicted cancers compared to traditional chemotherapy.
The MIT team is studying the chemistry behind KI-ARv-03’s selectivity as well as the mechanism of action for cell state disruption. They hope their findings will have broad applicability across many cancer types, particularly those whose malignancy is also sustained by oncogene addiction and dysregulated transcriptional programs.
“Prostate cancer is not unique in its resistance to treatment,” explains Koehler, “nor is it the only tumor type with a defined oncogene addiction. Studying the biochemical behavior/properties of KI-ARv-03 and comparing it to other compounds uncovered by our screens will help us generalize our findings to other cancer types and optimize development of other therapeutics.”
This work was supported, in part, by the Koch Institute-Dana-Farber/Harvard Cancer Center Bridge Project, the Koch Institute Support (core) Grant, the National Institutes of Health, the Royal G. and Mae H. Westaway Family Memorial Fund, the Ono Pharma Foundation, the MIT Center for Precision Cancer Medicine, and Janssen Pharmaceuticals, Inc., via the Transcend partnership with the Koch Institute.