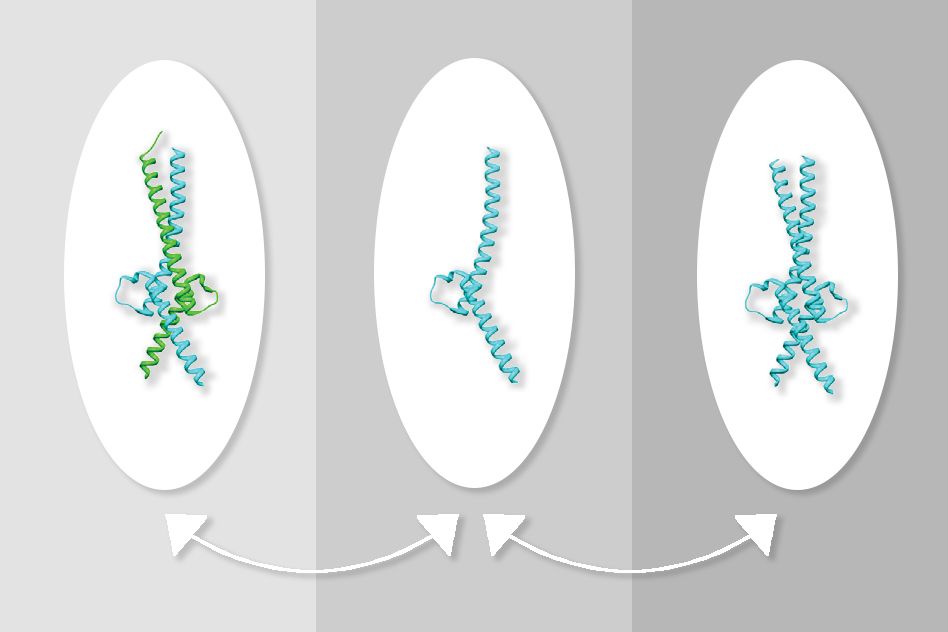
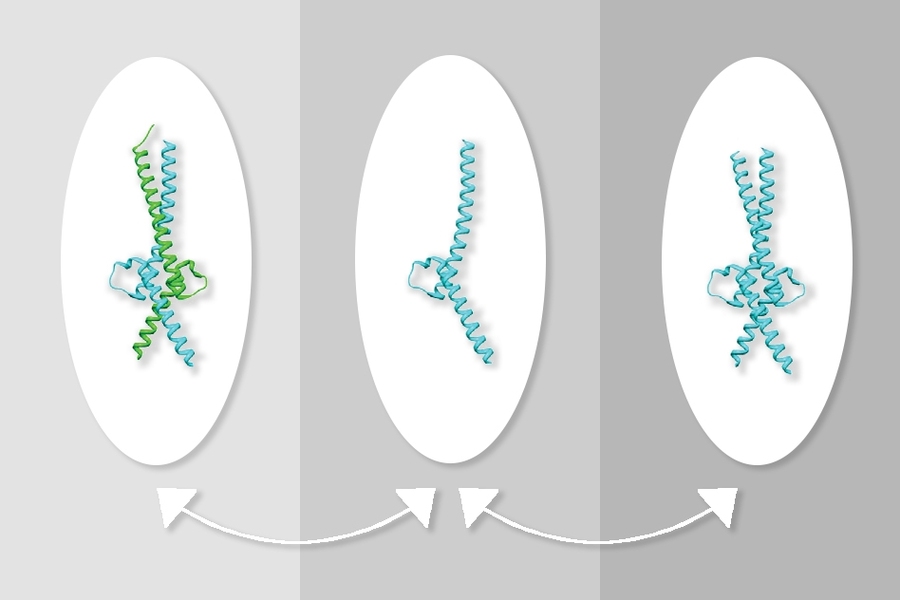
One of the most common cancer-promoting genes, known as Myc, is also one of the most difficult to target with drugs. Scientists have long tried to develop drugs that block the Myc protein, but so far their efforts have not been successful.
Now, using an alternative strategy, MIT researchers have discovered a compound that can reduce Myc activity by tying up the protein that is Myc’s usual binding partner, leaving Myc partnerless and unable to perform its usual functions.
The research team, led by Angela Koehler, an assistant professor of biological engineering and a member of MIT’s Koch Institute for Integrative Cancer Research, found that the compound they developed could suppress tumor growth in mice with certain types of cancer. The compound has been licensed by an MIT spinout that is now seeking to develop more powerful versions that could potentially be tested in human patients.
Koehler is the senior author of the study, which appears online in the journal Cell Chemical Biology on March 14. MIT postdoc Nicholas Struntz and graduate student Andrew Chen are the lead authors of the study, and the research team also includes authors from the Broad Institute of MIT and Harvard, Stanford University, Baylor College of Medicine, Brigham and Women’s Hospital, and Dana-Farber Cancer Institute.
A new approach
For decades, cancer researchers have been trying to find ways to shut off Myc, which is a transcription factor — a protein that controls the expression of other genes. Known as a “master regulator,” Myc controls many genes involved in basic cellular functions such as growth and metabolism. When it becomes overexpressed, as it does in about 70 percent of cancers, it drives uncontrolled cell growth and proliferation.
Myc usually forms a structure known as a heterodimer with the Max protein, and these proteins together bind to DNA to turn on gene transcription. Drug development efforts have traditionally focused on disrupting the interaction of Myc and Max, which has proven difficult. Most of the compounds that researchers have tested have proven too weak, or not specific enough to the Myc-Max interaction.
Koehler encountered similar difficulties, but several years ago, she decided to pursue a different strategy, based on the Max protein. The idea was to try to find compounds that would interact with Max, and then see if they had any effect on Myc’s ability to drive cell growth.
Using a technology developed by Koehler known as a microarray binding assay, the researchers screened a library of about 20,000 compounds, including both natural products and a collection of compounds synthesized by the Broad Institute, as possible drug candidates. The top six hits, in terms of ability to bind to Max and inhibit Myc transcriptional activity in another assay, all came from the Broad Institute collection.
The researchers tested the compounds in several different cancer cell lines and identified one that appeared to be most effective at halting cell growth.
At first, the researchers were unsure how this compound was blocking Myc activity, but experiments revealed that it was stabilizing a structure in which two molecules of Max bind together, forming a structure called a homodimer. This reduces the formation of the Myc-Max heterodimer and leads to a decrease in Myc levels, which the researchers believe may be the result of the unpartnered protein being broken down within cells.
Shrinking tumors
The researchers found that the compound slowed cell growth in a variety of Myc-dependent human cancer cells, including models for hepatocellular carcinoma, T-cell acute lymphoblastic leukemia, and Burkitt’s lymphoma.
They also tested the compound in mice, and found that even though the compound they originally identified was not optimized for maximum potency, it could slow tumor progression in mouse models of hepatocellular carcinoma and T-cell acute lymphoblastic leukemia.
“The discovery and detailed validation of a small molecule targeting Max homodimers represents a significant advance over previous attempts to directly inhibit either Myc itself or Myc-Max dimerization,” says Robert Eisenman, a principal investigator at the Fred Hutchinson Cancer Research Center, who was not involved in the study. “It not only provides new insight into how Myc functions but reveals what is likely to be an important exploitable vulnerability in Myc-driven cancers.”
Kronos Bio, the company that has licensed the rights to the compound described in this paper, is now working to optimize it to be more potent and more efficient. Koehler’s lab is also working on learning more about how this compound works, as well as determining the structure of the complex that it forms with the Max homodimer, in hopes of potentially developing better versions.
“This particular compound isn’t going to be a drug — it’s really just a tool to clarify the relevance of stabilizing Max homodimers as a strategy to perturb Myc function,” Koehler says. “That can guide people in the pharmaceutical industry who are thinking about trying to drug Myc, to maybe think about other ways to find Max homodimer stabilizers.”
Her lab is also pursuing other ways to target Myc, such as finding ways to stabilize a homodimer of two Myc molecules, which would likely end up being degraded within the cell.
“There may be different ways to stabilize biomolecular interactions within the Myc-Max network that could lead to different ways of perturbing Myc function,” she says.
The research was funded, in part, by the National Cancer Institute, including the Koch Institute Support (core) Grant, the National Institutes of Health, the Leukemia and Lymphoma Society, the Ono Pharma Foundation, the MIT Deshpande Center for Technological Innovation, the MIT Center for Precision Cancer Medicine, the AACR-Bayer Innovation and Discovery Grant, and the Merkin Institute Fellows Program at the Broad Institute.