For 30 years, the chemotherapy drug cisplatin has been one of doctors’ first lines of defense against tumors, especially those of the lung, ovary and testes. While cisplatin is often effective when first given, it has a major drawback: Tumors can become resistant to the drug and start growing again.
Now, MIT cancer biologists have shown how that resistance arises, a finding that could help researchers design new drugs that overcome cisplatin resistance. The team, led by Tyler Jacks, director of the David H. Koch Institute for Integrative Cancer Research at MIT, reports the results in the April 15 issue of the journal Genes and Development.
Cisplatin and other platinum-based cancer drugs destroy tumor cells by binding to DNA strands, interfering with DNA replication. That activates the cell’s DNA repair mechanisms, but if the damage is too extensive to be repaired, the cell undergoes programmed suicide.
Eventually, cancer cells learn to fight back. The new study shows that tumor cells treated with cisplatin ramp up their DNA repair pathways, allowing them to evade cell death, says Trudy Oliver, a postdoctoral fellow in Jacks’ lab and lead author of the paper.
Previous studies had suggested several possible mechanisms for resistance development, including enhancement of DNA repair pathways, detoxification of the drug, and changes in how the drug is imported into or exported out of the cell. However, those studies were done in cancer cells grown in the lab, not in living animals (in vivo).
“Many mechanisms have been identified but it’s not clear what happens in vivo because the in vivo environment is so much more complicated than in cell lines,” says Oliver.
Oliver and her colleagues set out to study cisplatin resistance in mice with a mutation in a gene called Kras, which leads the animals to develop lung cancer. About 30 percent of human lung cancer patients have mutations in Kras. Some of the mice also had defective versions of the tumor suppressor gene p53, which is mutated in about half of human lung cancers.
The researchers found that cisplatin was effective against lung tumors in both sets of mice, though it was more potent in mice that still had functional p53. In those mice, tumors actually shrank, while the drug only slowed tumor growth in mice with defective p53. Those results are consistent with findings in human patients.
After four doses of cisplatin, mice with normal p53 developed resistance to the drug, and tumors started growing faster. To figure out why, the researchers analyzed which genes were being transcribed more as resistance developed, and identified several that are involved in DNA repair pathways.
One gene that particularly caught the researchers’ attention is PIDD (p53-induced protein with a death domain), which is turned on by p53 and has been implicated in programmed cell death, though its exact function is not known. When PIDD levels are artificially increased in human lung cancer cells, they become more resistant to cisplatin. Oliver is now studying tumors in which the PIDD gene has been knocked out, to see if its absence hinders drug resistance.
It is likely that PIDD is just one of many genes, in many pathways, involved in the drug resistance process, says Oliver. “It’s not a simple phenomenon,” she says.
The work was funded by the National Institutes of Health and the National Cancer Institute.
This study represents an important step toward better understanding of how cisplatin resistance arises, says Thomas Helleday, professor of radiation oncology at the University of Oxford, who was not involved in the research. Helleday describes the work as a “landmark” paper that “has important implications in the design of new drugs, which should be targeted to stop the repair that is activated in cisplatin-resistant tumors.”
MIT biologists show how tumors can become resistant to the commonly used chemotherapy drug cisplatin.
Publication Date:
Caption:
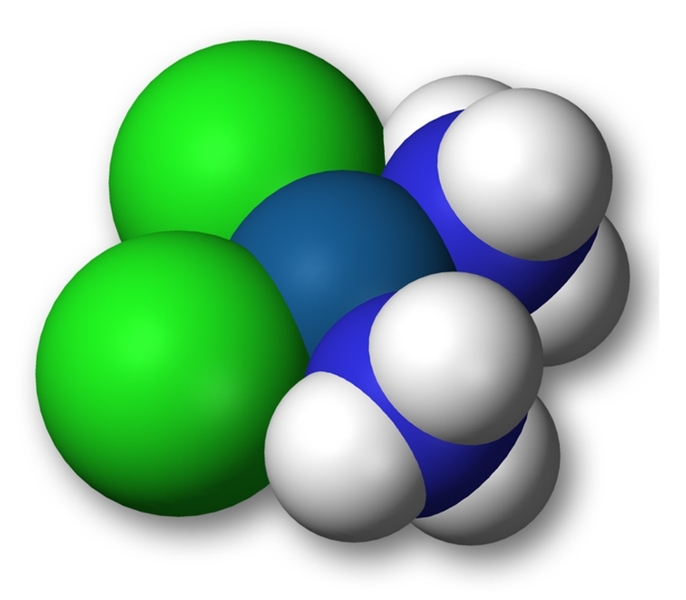
A 3-D model of a cisplatin molecule.

Caption:
Tyler Jacks, director of the David H. Koch Institute for Integrative Cancer Research at MIT.
Credits:
Photo: Kent Dayton Photography