Performance across a wide range of new technologies from solar cells to fuel cells depends on interactions at interfaces between materials on the atomic scale. “The behavior is influenced, and often dominated by, the interfaces,” says MIT assistant professor of mechanical engineering Alexie M. Kolpak.
Yet studies at the atomic scale — for example, at the interface between solid electrodes and water in a cell for splitting water (electrocatalysis) — are difficult to observe experimentally and costly to model computationally. Kolpak’s group at MIT is making progress in a number of different thrusts both through its own computational studies and through collaborations with experimentalists.
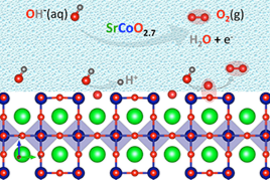
Kolpak, who is the Rockwell International Career Development Assistant Professor, and mechanical engineering doctoral student Xi (Jerry) Rong SM ’14 recently joined collaborators from the University of Texas at Austin and the Skoltech Center for Electrochemical Energy Storage to demonstrate the most efficient water-splitting catalyst, strontium cobaltite. Kolpak’s and Rong’s computational work showed the underlying phenomenon in this highly efficient catalyst is a newly understood mechanism, “lattice oxygen-mediated oxygen evolution reaction.”
Besides electrocatalysis, Kolpak and her team are also studying photocatalysis, solar cells, carbon dioxide (CO2) capture/conversion, and thermoelectric materials. “In all of these cases, the technology at this point is very much limited by our understanding of different interfaces, and if we can understand and control them, we can really start optimizing behavior in new ways,” Kolpak says. One powerful tool in this work is density functional theory (DFT), which enables computer-based modeling of atomic and electronic structure and properties.
Challenging “ideal” model
A standard assumption in modeling electrocatalytic reactions such as water-splitting has been to assume an ideal structure at the interface of the catalyst and the water. But, Kolpak’s recent work challenges that assumption. For one thing, the “lattice oxygen-mediated oxygen evolution reaction” analysis showed that oxygen atoms move into and out of the catalyst and alter its surface structure, with these structural phase changes controlling the elemental reactions that take place.
The joint project with experimentalists from the University of Texas at Austin and the Skoltech Center for Electrochemical Energy Storage came about through a Skoltech meeting in Moscow in June 2015. Keith J. Stevenson, one of the paper’s co-senior authors is director of the Skoltech Center for Electrochemical Energy Storage. “We both had some ideas for what the mechanism was, so we were able to explore more from first principles, and they were able to do more experiments so that we could put together the full story,” Kolpak says.
This new “lattice oxygen-mediated oxygen evolution reaction” (LOM) mechanism contrasts with prior assumptions that oxygen atoms in the perovskite metal oxide electrode did not themselves participate in the reaction. Stevenson first proposed direct participation of lattice hydroxide ions in alkaline oxygen evolution in a 2013 study of lanthanum nickel oxide.
Catalytic sweet spot
Changes in pH and electrical potential drive these surface structural changes. (The pH is alkaline in the case of these water reactions.) The impact is to alter the ratio of atomic elements and their spatial arrangement, which in turn alters electronic properties and catalytic activity. “Knowing what the surface structure is under certain conditions and then what the reactivity is on that surface structure can tell you where to go to get optimum performance of a particular material,” Kolpak says. The researchers also quantified the relationship between oxide stability and reactivity, identifying oxygen mobility as a key indicator for the efficiency of perovskite metal oxide catalysts. “This understanding of the relationship between the stability and the activity of the materials is really useful for saying where is that sweet spot between those two that will lead to highly active and long-lasting catalysts,” she explains.
Rong was lead author of a January 2016 ACS Catalysis paper with Kolpak and visiting scholar Jules Parolin in which they previously explored the new LOM mechanism, with the emphasis on its unprecedented correlation with oxide stability, for the rational design of catalysts. “Lattice oxygen participation is a prerequisite for the high efficiency of the catalyst, so how to tune the compositions, how to tune the properties, of the catalysts to be beneficial for this mechanism is the fundamental key step that people need to design catalysts in the future,” Rong says.
Eyeing larger systems
Beyond oxygen evolution and oxygen reduction, Kolpak is extending this approach to many different reactions, including CO2 reduction, conversion of ammonia, urea, and other compounds. “Another thing we’re trying to do is to really start studying the effect of having explicit water molecules in our calculations,” she says.
“We can add a couple of water molecules, or a layer of water molecules, and we’ve done that and seen, well it doesn’t really have much of an effect, but we don’t know if we’re missing something that happens when you have a whole continuum of water connected to the surface,” she explains.
To handle the larger universe of calculations for continuous water systems, as well as larger materials systems that incorporate grain boundaries and different defect configurations, Kolpak’s group is developing a machine-learning tool known as a neural network that processes thousands of individual density functional theory calculations into a coherent picture. “We’ll do a whole bunch of small [density functional theory] calculations with the atoms we’re interested in, in many different environments, then we use a neural network that will fit, or basically 'learn,' the potential energy surface that’s represented by all of these different configurations,” she says. Postdoc Brian Kolb and graduate student Levi Lentz are developing these neural network tools.
“The neural network actually learns the functional form of your data, so that’s very, very powerful, and you can get a very flexible potential that really is able to describe all these environments — of course depending on what you put into your reference data — so you can then use it to study large systems and perform calculations with many different configurations, all with the accuracy of DFT,” Kolpak says.
Water alters reactions
Kolpak used this neural network approach to show how the presence of water can change the outcome of chemical reactions in a 2014 Nano Letters paper on gold copper nanoparticles with postdoc Nong Artrith. “These nanoparticles are very well-studied, so people had seen previously that in vacuum you get gold at the surface, and when you put it in water, you get copper,” Kolpak explains. “We were able to find the composition, the stoichiometry, and the distribution of atoms that was thermodynamically favorable under given conditions, and we found that when we actually include water explicitly we get very, very different results from those you find using a typical approach, which would be to just look at the nanoparticles in vacuum. In vacuum, we have these very nice icosahedral nanoparticles with gold on the surface, and then in water, we find that we have much more spherical, lumpy nanoparticles, and the copper comes to the surface. ... But no one really had a good understanding of why, so it was nice that we could validate and explain the experimental results. More importantly, it showed that including the water molecules is actually very important in some systems,” Kolpak says.
The neural network computer coding has other functions as well. “What we fit is structure and energy to potential, so we get a potential energy surface that we can then use in a molecular dynamics calculation, but we can also fit things like structure to bandgap, or charge density to bandgap, or any combination of properties to some other property,” she says. “In principle, we can also use it to link up with experimentalists, so we can take data, for example, on processing conditions and if we had enough different samples and relate that to structure, then we can get relationships or mappings between processing parameters and the resulting structure. So this could be a really interesting way to link the processing parameters, the structure and the properties to the atomic and electronic scale that we study with DFT.”
Splitting excitons
Graduate student Levi Lentz is exploring the electronic, photovoltaic and thermoelectric properties of layered inorganic/organic hybrid materials known as 2-D transition metal phosphates. “We kind of discovered by just playing around with these materials that they actually have very interesting electronic transport properties, and further, that you can dope them,” Kolpak notes. These materials can be processed in solution. “They assemble and crystallize in solution, which would make them potentially very cheap.”
Lentz and Kolpak used density functional theory to design transition metal phosphate superlattices composed of alternating inorganic and organic layers that can split excitons and spatially separate the resulting electrons and holes. This means that in solar cell use, electrons could move along one layer and holes along a parallel layer, with no chance of recombining. Although this work is currently under review, Kolpak says, these materials hold promise for minimizing efficiency losses due to recombination while maximizing open circuit voltage in hybrid solar cells.
“We’re also now working in the context of Gang Chen’s S3TEC center on looking into if we can make these into good thermoelectric materials,” she adds. Studies are underway now to compute the thermal conductivity of these 2-D transition metal phosphates.
Life changing condition
Kolpak, 35, did not always dream of being a scientist. She was a competitive figure skater in her early years but turned to science in her teens after a diagnosis of rheumatoid arthritis forced her to give up skating. Her interest in ethics and genetic engineering in high school led her to study biochemistry at the University of Pennsylvania. There, she encountered quantum mechanics, and she was captivated. “The professor was Andrew Rappe, and I ended up doing my PhD with him in his group,” she recalls. Rappe PhD ’92 completed his doctoral studies at MIT with Francis Wright Davis Professor of Physics John D. Joannopoulos. Kolpak served as a postdoc at Yale University with professor of applied physics Sohrab Ismail-Beigi and at MIT with Department of Materials Science and Engineering Professor Jeffrey C. Grossman. Kolpak’s hobbies include painting with pastels and walking in the Back Bay Fens.
Putting classes online
During her first year on the MIT faculty in 2013, Kolpak worked with MIT mechanical engineering principal research scientist Simona Socrate to put a half-semester intro mechanics course, 2.01x (Elements of Structures) onto edX and also taught 2.02A (Engineering Materials). “Recently I’ve been teaching 2.087, which is Engineering Mathematics, so I’ve been teaching about linear algebra and ordinary differential equations,” she says.
Kolpak developed a graduate course at MIT in computational materials design. “I had a class that was mostly experimental students; they all learned how to do density functional theory calculations; and then they did a project using DFT that was related to some of their research. I had one student do a great project on the perovskite halides, the solar cell materials that everyone is excited about right now. That was really fun,” she recalls. Kolpak hopes to offer the course again, perhaps next spring.