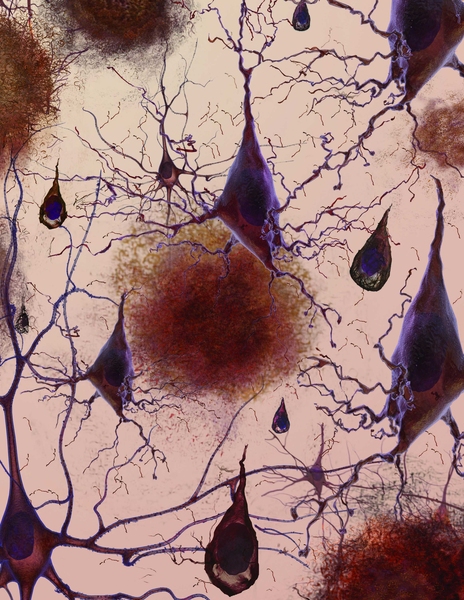
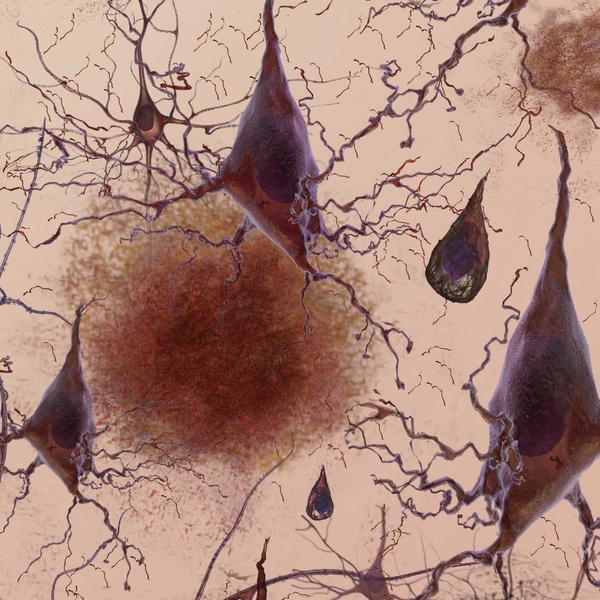
MIT neuroscientists have shown that an enzyme overproduced in the brains of Alzheimer’s patients creates a blockade that shuts off genes necessary to form new memories. Furthermore, by inhibiting that enzyme in mice, the researchers were able to reverse Alzheimer’s symptoms.
The finding suggests that drugs targeting the enzyme, known as HDAC2, could be a promising new approach to treating the disease, which affects 5.4 million Americans. The number of Alzheimer’s victims worldwide is expected to double every 20 years, and President Barack Obama recently set a target date of 2025 to find an effective treatment.
Li-Huei Tsai, leader of the research team, says that HDAC2 inhibitors could help achieve that goal, though it would likely take at least 10 years to develop and test such drugs.
“I would really strongly advocate for an active program to develop agents that can contain HDAC2 activity,” says Tsai, director of the Picower Institute for Learning and Memory at MIT. “The disease is so devastating and affects so many people, so I would encourage more people to think about this.”
Tsai and her colleagues report the findings in the Feb. 29 online edition of Nature. Lead author of the paper is Johannes Gräff, a postdoc at the Picower Institute.
Genome modification
Histone deacetylases (HDACs) are a family of 11 enzymes that control gene regulation by modifying histones — proteins around which DNA is spooled, forming a structure called chromatin. When HDACs alter a histone through a process called deacetylation, chromatin becomes more tightly packaged, making genes in that region less likely to be expressed.
HDAC inhibitors can reverse this effect, opening up the DNA and allowing it to be transcribed.
In previous studies, Tsai had shown that HDAC2 is a key regulator of learning and memory. In the new study, her team discovered that inhibiting HDAC2 can reverse Alzheimer’s symptoms in mice.
The researchers found that in mice with Alzheimer’s symptoms, HDAC2 (but not other HDACs) is overly abundant in the hippocampus, where new memories are formed. HDAC2 was most commonly found clinging to genes involved in synaptic plasticity — the brain’s ability to strengthen and weaken connections between neurons in response to new information, which is critical to forming memories. In the affected mice, those genes also had much lower levels of acetylation and expression.
“It’s not just one or two genes, it’s a group of genes that work in concert to control different phases of memory formation,” Tsai says. “With such a blockade, the brain really loses the ability to quickly respond to stimulation. You can imagine that this creates a huge problem in terms of learning and memory functions, and perhaps other cognitive functions.”
The researchers then shut off HDAC2 in the hippocampi of mice with Alzheimer’s symptoms, using a molecule called short hairpin RNA, which can be designed to bind to messenger RNA — the molecule that carries genetic instructions from DNA to the rest of the cell.
With HDAC2 activity reduced, histone acetylation resumed, allowing genes required for synaptic plasticity and other learning and memory processes to be expressed. In treated mice, synaptic density was greatly increased and the mice regained normal cognitive function.
“This result really advocates for the notion that if there is any agent that can selectively down-regulate HDAC2, it’s going to be very beneficial,” Tsai says.
The researchers also analyzed postmortem brains of Alzheimer’s patients and found elevated levels of HDAC2 in the hippocampus and entorhinal cortex, which play important roles in memory storage.
“What’s really valuable is that [Tsai] has identified which HDAC is involved, as well as delineated a pathway of how it can lead to impaired memory. It’s a really complete and well-executed study,” says Brett Langley, director of neuronal epigenetics at Burke Rehabilitation Center and assistant professor of neurology at Weill Cornell Medical School, who was not involved in this research.
Reversing the blockade
The findings may explain why drugs that clear beta-amyloid proteins from the brains of Alzheimer’s patients have offered only modest, if any, improvements in clinical trials, Tsai says.
Beta-amyloid proteins are known to clump in the brains of Alzheimer’s patients, interfering with a type of cell receptor needed for synaptic plasticity. The new study shows that beta amyloid also stimulates production of HDAC2, possibly initiating the blockade of learning and memory genes.
“We think that once this epigenetic blockade of gene expression is in place, clearing beta amyloid may not be sufficient to restore the active configuration of the chromatin,” Tsai says.
The appeal of HDAC2 inhibitors, Tsai says, is that they could conceivably reverse symptoms even after the blockade is well-established. However, much more drug development has to take place before such a compound could enter clinical trials. “It’s really hard to predict,” Tsai says. “Clinical trials would probably be five years down the line. And if everything goes well, to become an approved drug would probably take at least 10 years.”
Some general HDAC inhibitors, not specific to HDAC2, have been tested in clinical trials as cancer drugs. However, to treat Alzheimer’s, a more selective approach is needed, Tsai says. “You want something as selective as possible, and as safe as possible,” she says.
The finding suggests that drugs targeting the enzyme, known as HDAC2, could be a promising new approach to treating the disease, which affects 5.4 million Americans. The number of Alzheimer’s victims worldwide is expected to double every 20 years, and President Barack Obama recently set a target date of 2025 to find an effective treatment.
Li-Huei Tsai, leader of the research team, says that HDAC2 inhibitors could help achieve that goal, though it would likely take at least 10 years to develop and test such drugs.
“I would really strongly advocate for an active program to develop agents that can contain HDAC2 activity,” says Tsai, director of the Picower Institute for Learning and Memory at MIT. “The disease is so devastating and affects so many people, so I would encourage more people to think about this.”
Tsai and her colleagues report the findings in the Feb. 29 online edition of Nature. Lead author of the paper is Johannes Gräff, a postdoc at the Picower Institute.
Genome modification
Histone deacetylases (HDACs) are a family of 11 enzymes that control gene regulation by modifying histones — proteins around which DNA is spooled, forming a structure called chromatin. When HDACs alter a histone through a process called deacetylation, chromatin becomes more tightly packaged, making genes in that region less likely to be expressed.
HDAC inhibitors can reverse this effect, opening up the DNA and allowing it to be transcribed.
In previous studies, Tsai had shown that HDAC2 is a key regulator of learning and memory. In the new study, her team discovered that inhibiting HDAC2 can reverse Alzheimer’s symptoms in mice.
The researchers found that in mice with Alzheimer’s symptoms, HDAC2 (but not other HDACs) is overly abundant in the hippocampus, where new memories are formed. HDAC2 was most commonly found clinging to genes involved in synaptic plasticity — the brain’s ability to strengthen and weaken connections between neurons in response to new information, which is critical to forming memories. In the affected mice, those genes also had much lower levels of acetylation and expression.
“It’s not just one or two genes, it’s a group of genes that work in concert to control different phases of memory formation,” Tsai says. “With such a blockade, the brain really loses the ability to quickly respond to stimulation. You can imagine that this creates a huge problem in terms of learning and memory functions, and perhaps other cognitive functions.”
The researchers then shut off HDAC2 in the hippocampi of mice with Alzheimer’s symptoms, using a molecule called short hairpin RNA, which can be designed to bind to messenger RNA — the molecule that carries genetic instructions from DNA to the rest of the cell.
With HDAC2 activity reduced, histone acetylation resumed, allowing genes required for synaptic plasticity and other learning and memory processes to be expressed. In treated mice, synaptic density was greatly increased and the mice regained normal cognitive function.
“This result really advocates for the notion that if there is any agent that can selectively down-regulate HDAC2, it’s going to be very beneficial,” Tsai says.
The researchers also analyzed postmortem brains of Alzheimer’s patients and found elevated levels of HDAC2 in the hippocampus and entorhinal cortex, which play important roles in memory storage.
“What’s really valuable is that [Tsai] has identified which HDAC is involved, as well as delineated a pathway of how it can lead to impaired memory. It’s a really complete and well-executed study,” says Brett Langley, director of neuronal epigenetics at Burke Rehabilitation Center and assistant professor of neurology at Weill Cornell Medical School, who was not involved in this research.
Reversing the blockade
The findings may explain why drugs that clear beta-amyloid proteins from the brains of Alzheimer’s patients have offered only modest, if any, improvements in clinical trials, Tsai says.
Beta-amyloid proteins are known to clump in the brains of Alzheimer’s patients, interfering with a type of cell receptor needed for synaptic plasticity. The new study shows that beta amyloid also stimulates production of HDAC2, possibly initiating the blockade of learning and memory genes.
“We think that once this epigenetic blockade of gene expression is in place, clearing beta amyloid may not be sufficient to restore the active configuration of the chromatin,” Tsai says.
The appeal of HDAC2 inhibitors, Tsai says, is that they could conceivably reverse symptoms even after the blockade is well-established. However, much more drug development has to take place before such a compound could enter clinical trials. “It’s really hard to predict,” Tsai says. “Clinical trials would probably be five years down the line. And if everything goes well, to become an approved drug would probably take at least 10 years.”
Some general HDAC inhibitors, not specific to HDAC2, have been tested in clinical trials as cancer drugs. However, to treat Alzheimer’s, a more selective approach is needed, Tsai says. “You want something as selective as possible, and as safe as possible,” she says.