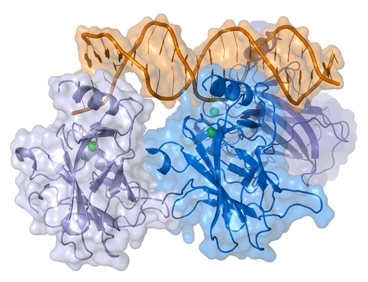
Since the early 1980s, cancer researchers have known that a protein called p53 plays a critical role in protecting cells from becoming cancerous. The protein is defective in about half of all human cancers; when it functions correctly, it appears to suppress tumor formation by preventing cells with cancer-promoting mutations from reproducing.
Knowing p53’s critical role in controlling cancer, researchers have been trying to develop drugs that restore the protein’s function, in hopes of re-establishing the ability to suppress tumor growth. One such drug is now in clinical trials.
In a new study that highlights a possible limitation of such drugs, MIT cancer biologists show that restoring p53’s function in mice with lung cancer has no effect early in tumor development, but restoring the function later on could prevent more advanced tumors from spreading throughout the body.
The findings, reported in the Nov. 25 issue of Nature, suggest that drugs that restore p53 function could help prevent aggressive lung cancers from metastasizing, though they might spare benign tumor cells that could later turn aggressive. “Even if you clear the malignant cells, you’re still left with benign cells harboring the p53 mutation,” says David Feldser, lead author of the paper and a postdoctoral fellow at the David H. Koch Institute for Integrative Cancer Research at MIT.
However, such drugs are still worth pursuing because they could prolong the life of the patient, says Feldser, who works in the lab of Koch Institute Director Tyler Jacks, senior author of the paper. The research was funded by the Howard Hughes Medical Institute.
Guardian of the genome
The p53 protein is known to control the cell cycle, which regulates cell division. In particular, the protein stops a cell from dividing when its DNA is damaged. It then activates DNA repair systems, and if the damage proves irreparable, it instructs the cell to commit suicide.
Without p53, cells can continue dividing even after acquiring hazardous mutations. Eventually, after a cell accumulates enough mutations, it becomes cancerous. Cancer biologists believe that sustained inactivation of p53 and other tumor suppressors is necessary for cancers to become advanced.
In the new Nature study, the MIT researchers studied mice that are genetically engineered to develop lung tumors shortly after birth. Those mice also have an inactive form of the p53 gene, but the gene includes a genetic "switch" that allows the researchers to turn it back on after tumors develop.
At first, the researchers turned on p53 in mice that were four weeks old and had developed tumors known as adenomas, which are benign. To their surprise, restoring p53 had no effect on the tumors.
Next they turned on p53 in another group of tumor-prone mice, but they waited until the mice were 10 weeks old. At this point, their tumors had progressed to adenocarcinomas, a malignant type of cancer. In these mice, turning on p53 cleared the malignant cells, but left behind cells that had not become malignant.
This suggests that the p53 signaling pathway is recruited only when there is a lot of activity from other cancer genes. In benign tumors, there is not enough activity to engage the p53 system, so restoring it has no effect on those tumors. In the malignant tumor cells, reactivated p53 eliminates cells with too much activity in a signaling pathway involving mitogen-activated protein kinase (MAPK), which is often overactive in cancer cells, leading to uncontrolled growth.
In this study, the researchers restored normal levels of p53, but a p53-activating drug would likely generate much higher levels of the protein, says Geoffrey Wahl, a professor at the Salk Institute. With elevated p53, “it might be that you get a more significant response than what was observed here,” he says.
Wahl, who was not involved in this research, says the study sounds a cautionary note about the levels of p53 needed to effectively clear a tumor. “If you don’t have adequate p53 activation, it’s going to allow some cancer cells to escape,” he says.
The MIT researchers are now looking for drugs that reactivate mutant forms of p53, and also plan to study whether tumors that have metastasized would be vulnerable to p53 restoration.
Knowing p53’s critical role in controlling cancer, researchers have been trying to develop drugs that restore the protein’s function, in hopes of re-establishing the ability to suppress tumor growth. One such drug is now in clinical trials.
In a new study that highlights a possible limitation of such drugs, MIT cancer biologists show that restoring p53’s function in mice with lung cancer has no effect early in tumor development, but restoring the function later on could prevent more advanced tumors from spreading throughout the body.
The findings, reported in the Nov. 25 issue of Nature, suggest that drugs that restore p53 function could help prevent aggressive lung cancers from metastasizing, though they might spare benign tumor cells that could later turn aggressive. “Even if you clear the malignant cells, you’re still left with benign cells harboring the p53 mutation,” says David Feldser, lead author of the paper and a postdoctoral fellow at the David H. Koch Institute for Integrative Cancer Research at MIT.
However, such drugs are still worth pursuing because they could prolong the life of the patient, says Feldser, who works in the lab of Koch Institute Director Tyler Jacks, senior author of the paper. The research was funded by the Howard Hughes Medical Institute.
Guardian of the genome
The p53 protein is known to control the cell cycle, which regulates cell division. In particular, the protein stops a cell from dividing when its DNA is damaged. It then activates DNA repair systems, and if the damage proves irreparable, it instructs the cell to commit suicide.
Without p53, cells can continue dividing even after acquiring hazardous mutations. Eventually, after a cell accumulates enough mutations, it becomes cancerous. Cancer biologists believe that sustained inactivation of p53 and other tumor suppressors is necessary for cancers to become advanced.
In the new Nature study, the MIT researchers studied mice that are genetically engineered to develop lung tumors shortly after birth. Those mice also have an inactive form of the p53 gene, but the gene includes a genetic "switch" that allows the researchers to turn it back on after tumors develop.
At first, the researchers turned on p53 in mice that were four weeks old and had developed tumors known as adenomas, which are benign. To their surprise, restoring p53 had no effect on the tumors.
Next they turned on p53 in another group of tumor-prone mice, but they waited until the mice were 10 weeks old. At this point, their tumors had progressed to adenocarcinomas, a malignant type of cancer. In these mice, turning on p53 cleared the malignant cells, but left behind cells that had not become malignant.
This suggests that the p53 signaling pathway is recruited only when there is a lot of activity from other cancer genes. In benign tumors, there is not enough activity to engage the p53 system, so restoring it has no effect on those tumors. In the malignant tumor cells, reactivated p53 eliminates cells with too much activity in a signaling pathway involving mitogen-activated protein kinase (MAPK), which is often overactive in cancer cells, leading to uncontrolled growth.
In this study, the researchers restored normal levels of p53, but a p53-activating drug would likely generate much higher levels of the protein, says Geoffrey Wahl, a professor at the Salk Institute. With elevated p53, “it might be that you get a more significant response than what was observed here,” he says.
Wahl, who was not involved in this research, says the study sounds a cautionary note about the levels of p53 needed to effectively clear a tumor. “If you don’t have adequate p53 activation, it’s going to allow some cancer cells to escape,” he says.
The MIT researchers are now looking for drugs that reactivate mutant forms of p53, and also plan to study whether tumors that have metastasized would be vulnerable to p53 restoration.