The blobfish, once considered the ugliest animal in the world, has since had quite the redemption arc. Years after it was first discovered, scientists realized that the deep-sea creature appeared so unnervingly blobby only because it went through an extreme change in pressure when it was brought up to the surface. In its natural environment, 4,000 feet underwater, the fish looks perfectly handsome.
Structural biologists, whose goal is to deduce a molecule’s structure and function within a cell, face the risk of making a similar mistake. If biomolecular complexes are extracted from the cell, better-quality images can be obtained, but the molecules may not look natural. On the other hand, studying molecules without disrupting their environment at all is technically challenging, like filming deep underwater.
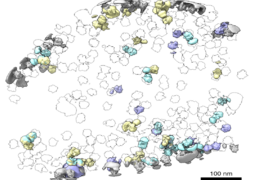
A new method, called purification-free ribosome imaging from subcellular mixtures (cryoPRISM), offers an appealing compromise. Developed by graduate students Mira May and Gabriela López-Pérez in the Davis lab in the MIT Department of Biology and recently published in PNAS, the technique allows biologists to visualize molecular complexes without taking them too far out of their natural context.
CryoPRISM captures molecular structures in cells that have just been broken open. This comes as close to preserving the natural interactions between molecules as possible, short of the extremely resource-intensive in-cell structural imaging, according to associate professor of biology Joey Davis, the faculty lead of the study.
“We think that the cryoPRISM method is a sweet spot where we preserve much of the native cellular contacts, but still have the resolution that lets us actually see molecular details,” Davis says. “Even in the extremely well-trodden system of translation in E. coli, which people have worked on for over 50 years, we are still finding new states that had just escaped people’s attention.”
A negative control that was not so negative
The development of cryoPRISM, as many discoveries in science, resulted from an unexpected observation that Mira May, the co-first author of the study, made while working on a different project.
Like all living organisms, bacteria rely on a process called translation to manufacture the proteins that carry out essential functions within the cell, from copying DNA to digesting nutrients. A key machine involved in translation is the ribosome — a biomolecular complex that assembles proteins based on instructions encoded by another molecule called mRNA. To regulate its activity, cells employ additional proteins that can change the shape of the ribosome, thus guiding its function.
May sought to identify new players in ribosomal regulation using cryoEM, by rapidly freezing lots of purified molecules and collecting thousands of 2D images to reconstruct their 3D structures. May was trying to pull ribosomes out of cells to visualize them together with their regulators. For her experiments, she designed a negative control containing unpurified bacterial lysate — a mixture of everything spilled from burst cells.
May expected to get noisy, low-quality images from this sample. To her surprise, instead, she saw intact ribosomes together with their natural interacting partners.
In just a few days, this technique experimentally validated data that would have taken months to acquire using other approaches.
“As I found more and more ribosomal states, this project became a method, not just a one-off finding,” May recalls.
Discovering new biology in a saturated field
Once May and her colleagues were confident that cryoPRISM could detect known ribosomal states, they began searching for ones that had previously escaped detection.
“It’s not just that we can recapitulate things that have been previously observed, but we can actually also discover novel ribosomal biology,” May says.
One of the novel states May identified has important implications for our understanding of the evolution of translation regulation.
During active translation, bacterial ribosomes are accompanied by a group of helper proteins called elongation factors. These factors bring in the materials for protein synthesis, like tRNAs and amino acids.
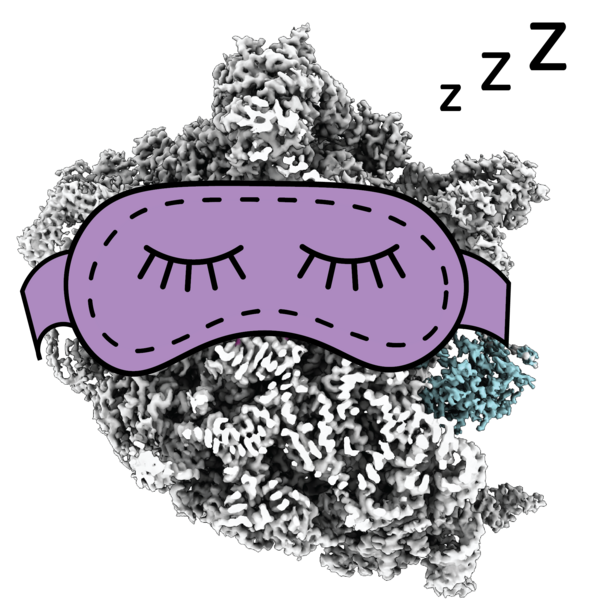
When cells encounter unfavorable conditions, such as colder temperatures, they reduce translation, which means that many ribosomes are out of work. These idle, hibernating ribosomes stop decoding mRNA, and the interface where they usually interact with helper molecules gets blocked by a hibernation factor called RaiA. This protein helps idle ribosomes avoid reactivation, like a sleeping mask that prevents a person from being woken up by light.
May observed the idle ribosomal state in her data, which on its own did not surprise her – this state had been described before. What surprised her was that some inactive ribosomes were interacting not only with RaiA, but also with an elongation factor called EF-G, which in bacteria was previously believed to only interact with active ribosomes.
A similar phenomenon has been seen before in more complex organisms, but observing it in a microbe suggests that its evolutionary origin may be older than previously thought.
“It fits an emerging model in the field, that elongation factors might bind to hibernating ribosomes to protect both the ribosome and themselves from degradation during periods of stress,” May explains. “Think of it like short-term storage.”
An unstressed cell might quickly eliminate unneeded inactive ribosomes, but because any stressor that puts ribosomes to sleep could be temporary, the cell may prefer to hold off on destroying them. That way, the ribosomes can be quickly reactivated if conditions improve.
The future of cryoPRISM
May has already teamed up with other MIT researchers to use cryoPRISM to visualize ribosomes in cells that are notoriously difficult to work with, including pathogenic organisms, which can be challenging to culture at the scale required for particle purification, and red blood cells isolated from patients, which cannot be cultured at all.
Besides its immediate application for translation research, cryoPRISM is a stepping stone toward the broader goal of structural biology: studying biomolecules in their natural environment.
To truly learn about deep-sea fish, scientists need to look at them in the deep sea; and to learn about cellular machines, scientists need to look at them in cells. According to Davis, cryoPRISM perfectly fits into the “theme of structural biology moving closer and closer to cellular context.”
This work was carried out, in part, with the use of MIT.nano facilities.