With the release of platforms like DALL-E 2 and Midjourney, diffusion generative models have achieved mainstream popularity, owing to their ability to generate a series of absurd, breathtaking, and often meme-worthy images from text prompts like “teddy bears working on new AI research on the moon in the 1980s.” But a team of researchers at MIT's Abdul Latif Jameel Clinic for Machine Learning in Health (Jameel Clinic) thinks there could be more to diffusion generative models than just creating surreal images — they could accelerate the development of new drugs and reduce the likelihood of adverse side effects.
A paper introducing this new molecular docking model, called DiffDock, will be presented at the 11th International Conference on Learning Representations. The model's unique approach to computational drug design is a paradigm shift from current state-of-the-art tools that most pharmaceutical companies use, presenting a major opportunity for an overhaul of the traditional drug development pipeline.
Drugs typically function by interacting with the proteins that make up our bodies, or proteins of bacteria and viruses. Molecular docking was developed to gain insight into these interactions by predicting the atomic 3D coordinates with which a ligand (i.e., drug molecule) and protein could bind together.
While molecular docking has led to the successful identification of drugs that now treat HIV and cancer, with each drug averaging a decade of development time and 90 percent of drug candidates failing costly clinical trials (most studies estimate average drug development costs to be around $1 billion to over $2 billion per drug), it’s no wonder that researchers are looking for faster, more efficient ways to sift through potential drug molecules.
Currently, most molecular docking tools used for in-silico drug design take a “sampling and scoring” approach, searching for a ligand “pose” that best fits the protein pocket. This time-consuming process evaluates a large number of different poses, then scores them based on how well the ligand binds to the protein.
In previous deep-learning solutions, molecular docking is treated as a regression problem. In other words, “it assumes that you have a single target that you’re trying to optimize for and there’s a single right answer,” says Gabriele Corso, co-author and second-year MIT PhD student in electrical engineering and computer science who is an affiliate of the MIT Computer Sciences and Artificial Intelligence Laboratory (CSAIL). “With generative modeling, you assume that there is a distribution of possible answers — this is critical in the presence of uncertainty.”
“Instead of a single prediction as previously, you now allow multiple poses to be predicted, and each one with a different probability,” adds Hannes Stärk, co-author and first-year MIT PhD student in electrical engineering and computer science who is an affiliate of the MIT Computer Sciences and Artificial Intelligence Laboratory (CSAIL). As a result, the model doesn't need to compromise in attempting to arrive at a single conclusion, which can be a recipe for failure.
To understand how diffusion generative models work, it is helpful to explain them based on image-generating diffusion models. Here, diffusion models gradually add random noise to a 2D image through a series of steps, destroying the data in the image until it becomes nothing but grainy static. A neural network is then trained to recover the original image by reversing this noising process. The model can then generate new data by starting from a random configuration and iteratively removing the noise.
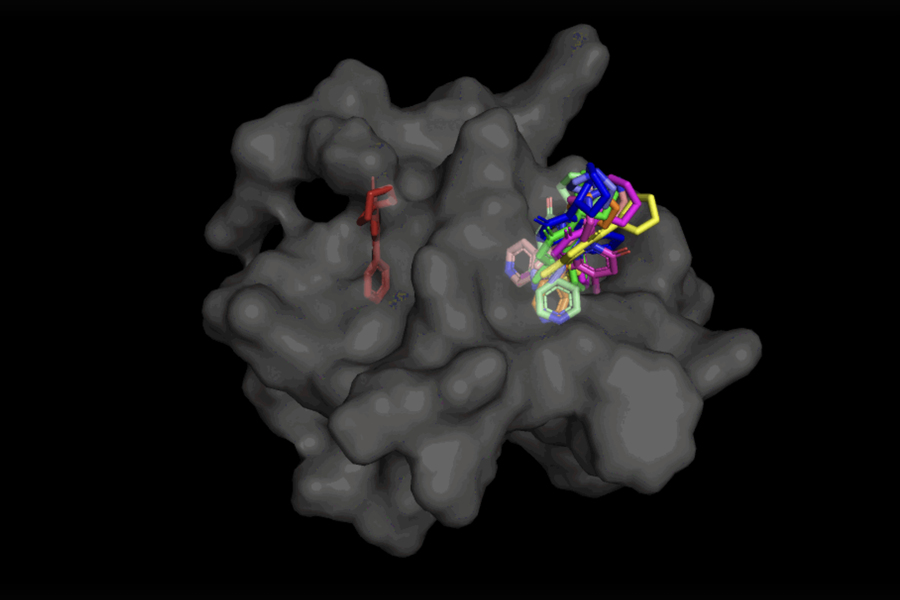
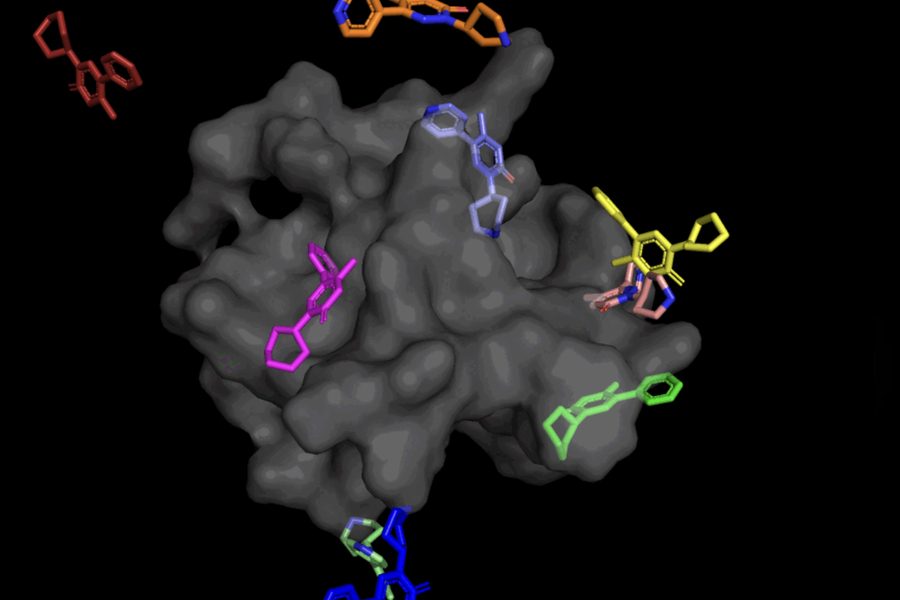
In the case of DiffDock, after being trained on a variety of ligand and protein poses, the model is able to successfully identify multiple binding sites on proteins that it has never encountered before. Instead of generating new image data, it generates new 3D coordinates that help the ligand find potential angles that would allow it to fit into the protein pocket.
This “blind docking” approach creates new opportunities to take advantage of AlphaFold 2 (2020), DeepMind’s famous protein folding AI model. Since AlphaFold 1’s initial release in 2018, there has been a great deal of excitement in the research community over the potential of AlphaFold’s computationally folded protein structures to help identify new drug mechanisms of action. But state-of-the-art molecular docking tools have yet to demonstrate that their performance in binding ligands to computationally predicted structures is any better than random chance.
Not only is DiffDock significantly more accurate than previous approaches to traditional docking benchmarks, thanks to its ability to reason at a higher scale and implicitly model some of the protein flexibility, DiffDock maintains high performance, even as other docking models begin to fail. In the more realistic scenario involving the use of computationally generated unbound protein structures, DiffDock places 22 percent of its predictions within 2 angstroms (widely considered to be the threshold for an accurate pose, 1Å corresponds to one over 10 billion meters), more than double other docking models barely hovering over 10 percent for some and dropping as low as 1.7 percent.
These improvements create a new landscape of opportunities for biological research and drug discovery. For instance, many drugs are found via a process known as phenotypic screening, in which researchers observe the effects of a given drug on a disease without knowing which proteins the drug is acting upon. Discovering the mechanism of action of the drug is then critical to understanding how the drug can be improved and its potential side effects. This process, known as “reverse screening,” can be extremely challenging and costly, but a combination of protein folding techniques and DiffDock may allow performing a large part of the process in silico, allowing potential “off-target” side effects to be identified early on before clinical trials take place.
“DiffDock makes drug target identification much more possible. Before, one had to do laborious and costly experiments (months to years) with each protein to define the drug docking. But now, one can screen many proteins and do the triaging virtually in a day,” Tim Peterson, an assistant professor at the University of Washington St. Louis School of Medicine, says. Peterson used DiffDock to characterize the mechanism of action of a novel drug candidate treating aging-related diseases in a recent paper. “There is a very ‘fate loves irony’ aspect that Eroom’s law — that drug discovery takes longer and costs more money each year — is being solved by its namesake Moore’s law — that computers get faster and cheaper each year — using tools such as DiffDock.”
This work was conducted by MIT PhD students Gabriele Corso, Hannes Stärk, and Bowen Jing, and their advisors, Professor Regina Barzilay and Professor Tommi Jaakkola, and was supported by the Machine Learning for Pharmaceutical Discovery and Synthesis consortium, the Jameel Clinic, the DTRA Discovery of Medical Countermeasures Against New and Emerging Threats program, the DARPA Accelerated Molecular Discovery program, the Sanofi Computational Antibody Design grant, and a Department of Energy Computational Science Graduate Fellowship.