Amyotrophic lateral sclerosis (ALS) — also known as Lou Gehrig’s disease — is a neurodegenerative disease that destroys the neurons that control muscle movement. There is no cure for ALS, which kills most patients within three to five years of the onset of symptoms, and about 5,600 new cases are diagnosed in the United States each year.
MIT neuroscientists have found new evidence that suggests that a failure to repair damaged DNA could underlie not only ALS, but also other neurodegenerative disorders such as Alzheimer’s disease. These findings imply that drugs that bolster neurons’ DNA-repair capacity could help ALS patients, says Li-Huei Tsai, director of MIT’s Picower Institute for Learning and Memory and senior author of a paper describing the ALS findings in the Sept. 15 issue of Nature Neuroscience.
Neurons are some of the longest-living cells in the human body. While other cells are frequently replaced, our neurons are generally retained throughout our lifetimes. Consequently, neurons can accrue a lot of DNA damage and are especially vulnerable to its effects.
“Our genome is constantly under attack and DNA strand breaks are produced all the time. Fortunately, they are not a worry because we have the machinery to repair it right away. But if this repair machinery were to somehow become compromised, then it could be very devastating for neurons,” Tsai says.
Lead authors of the paper are Picower Institute postdoc Wen-Yuan Wang and research scientist Ling Pan.
Impaired repair
Tsai’s group has been interested in understanding the importance of DNA repair in neurodegenerative processes for several years. In a study published in 2008, they reported that DNA double-strand breaks precede neuronal loss in a mouse model that undergoes Alzheimer’s disease-like neurodegeneration and identified a protein, HDAC1, which prevents neuronal loss under these conditions.
HDAC1 is a histone deacetylase, an enzyme that regulates genes by modifying chromatin, which consists of DNA wrapped around a core of proteins called histones. HDAC1 activity normally causes DNA to wrap more tightly around histones, preventing gene expression. However, it turns out that cells, including neurons, also exploit HDAC1’s ability to tighten up chromatin to stabilize broken DNA ends and promote their repair.
In a paper published earlier this year in Nature Neuroscience, Tsai’s team reported that HDAC1 works cooperatively with another deacetylase called SIRT1 to repair DNA and prevent the accumulation of damage that could promote neurodegeneration.
When a neuron suffers double-strand breaks, SIRT1 migrates within seconds to the damaged sites, where it soon recruits HDAC1 and other repair factors. SIRT1 also stimulates the enzymatic activity of HDAC1, which allows the broken DNA ends to be resealed.
SIRT1 itself has recently gained notoriety as the protein that promotes longevity and protects against diseases including diabetes and Alzheimer’s disease, and Tsai’s group believes that its role in DNA repair contributes significantly to the protective effects of SIRT1.
In an attempt to further unveil other partners that work with HDAC1 to repair DNA, Tsai and colleagues stumbled upon a protein called Fused In Sarcoma (FUS). This finding was intriguing, Tsai says, because the FUS gene is one of the most common sites of mutations that cause inherited forms of ALS.
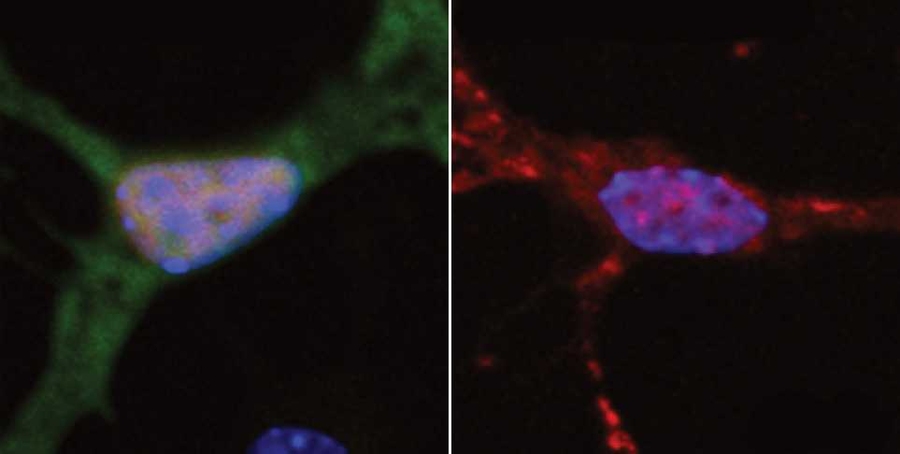
The MIT team found that FUS appears at the scene of DNA damage very rapidly, suggesting that FUS is orchestrating the repair response. One of its roles is to recruit HDAC1 to the DNA damage site. Without it, HDAC1 does not appear and the necessary repair does not occur. Tsai believes that FUS may also be involved in sensing when DNA damage has occurred.
Linking mutation and disease
At least 50 mutations in the FUS gene have been found to cause ALS. The majority of these mutations occur in two sections of the FUS protein. The MIT team mapped the interactions between FUS and HDAC1 and found that these same two sections of the FUS protein bind to HDAC1.
They also generated four FUS mutants that are most commonly seen in ALS patients. When they replaced the normal FUS with these mutants, they found that the interaction with HDAC1 was impaired and DNA damage was significantly increased. This suggests that those mutations prevent FUS from recruiting HDAC1 when DNA damage occurs, allowing damage to accumulate and eventually leading to ALS.
The researchers also analyzed brain tissue samples from ALS patients harboring FUS mutations and found that the amount of DNA damage in neurons in motor cortex was about double that found in normal brain tissue.
ALS patients with FUS mutations usually develop the disease early, before age 40. Only one of a person’s two copies of the FUS gene needs to be mutated to produce the disease. Tsai says that early in life, having one copy of the normal FUS gene may be enough to keep DNA repair going. “With aging, eventually the machinery is compromised and it contributes to neuronal demise,” she says.
The findings suggest that drugs that promote DNA damage repair, including activators of HDAC1 and SIRT1, could help combat the effects of ALS. SIRT1 activators are now being developed and have entered clinical trials to treat diabetes.
“There are numerous human inherited DNA-repair deficiency syndromes, many of which show neurodegeneration or other neurological defects. This new study now extends the spectrum of neuropathology caused by defects in DNA maintenance to include ALS,” says Peter McKinnon, a professor of genetics at St. Jude Children’s Research Hospital who was not part of the research team. “This study offers new avenues to explore in the quest for treatment strategies.”
Tsai’s lab is now studying whether there is a direct relationship between FUS and SIRT1. She also wants to determine whether the DNA damage that occurs in ALS patients after FUS is lost occurs in certain “hotspots” or is random. “I would speculate that there’s got to be hotspots in terms of where the DNA is damaged. But right now it remains speculation,” she says. “We really need to do the experiments and demonstrate whether that’s the case.”
The research was funded by the Simons Foundation, the National Institutes of Health, the Howard Hughes Medical Institute, the U.S. Department of Veteran Affairs, and the Muscular Dystrophy Association.
MIT neuroscientists have found new evidence that suggests that a failure to repair damaged DNA could underlie not only ALS, but also other neurodegenerative disorders such as Alzheimer’s disease. These findings imply that drugs that bolster neurons’ DNA-repair capacity could help ALS patients, says Li-Huei Tsai, director of MIT’s Picower Institute for Learning and Memory and senior author of a paper describing the ALS findings in the Sept. 15 issue of Nature Neuroscience.
Neurons are some of the longest-living cells in the human body. While other cells are frequently replaced, our neurons are generally retained throughout our lifetimes. Consequently, neurons can accrue a lot of DNA damage and are especially vulnerable to its effects.
“Our genome is constantly under attack and DNA strand breaks are produced all the time. Fortunately, they are not a worry because we have the machinery to repair it right away. But if this repair machinery were to somehow become compromised, then it could be very devastating for neurons,” Tsai says.
Lead authors of the paper are Picower Institute postdoc Wen-Yuan Wang and research scientist Ling Pan.
Impaired repair
Tsai’s group has been interested in understanding the importance of DNA repair in neurodegenerative processes for several years. In a study published in 2008, they reported that DNA double-strand breaks precede neuronal loss in a mouse model that undergoes Alzheimer’s disease-like neurodegeneration and identified a protein, HDAC1, which prevents neuronal loss under these conditions.
HDAC1 is a histone deacetylase, an enzyme that regulates genes by modifying chromatin, which consists of DNA wrapped around a core of proteins called histones. HDAC1 activity normally causes DNA to wrap more tightly around histones, preventing gene expression. However, it turns out that cells, including neurons, also exploit HDAC1’s ability to tighten up chromatin to stabilize broken DNA ends and promote their repair.
In a paper published earlier this year in Nature Neuroscience, Tsai’s team reported that HDAC1 works cooperatively with another deacetylase called SIRT1 to repair DNA and prevent the accumulation of damage that could promote neurodegeneration.
When a neuron suffers double-strand breaks, SIRT1 migrates within seconds to the damaged sites, where it soon recruits HDAC1 and other repair factors. SIRT1 also stimulates the enzymatic activity of HDAC1, which allows the broken DNA ends to be resealed.
SIRT1 itself has recently gained notoriety as the protein that promotes longevity and protects against diseases including diabetes and Alzheimer’s disease, and Tsai’s group believes that its role in DNA repair contributes significantly to the protective effects of SIRT1.
In an attempt to further unveil other partners that work with HDAC1 to repair DNA, Tsai and colleagues stumbled upon a protein called Fused In Sarcoma (FUS). This finding was intriguing, Tsai says, because the FUS gene is one of the most common sites of mutations that cause inherited forms of ALS.
The MIT team found that FUS appears at the scene of DNA damage very rapidly, suggesting that FUS is orchestrating the repair response. One of its roles is to recruit HDAC1 to the DNA damage site. Without it, HDAC1 does not appear and the necessary repair does not occur. Tsai believes that FUS may also be involved in sensing when DNA damage has occurred.
Linking mutation and disease
At least 50 mutations in the FUS gene have been found to cause ALS. The majority of these mutations occur in two sections of the FUS protein. The MIT team mapped the interactions between FUS and HDAC1 and found that these same two sections of the FUS protein bind to HDAC1.
They also generated four FUS mutants that are most commonly seen in ALS patients. When they replaced the normal FUS with these mutants, they found that the interaction with HDAC1 was impaired and DNA damage was significantly increased. This suggests that those mutations prevent FUS from recruiting HDAC1 when DNA damage occurs, allowing damage to accumulate and eventually leading to ALS.
The researchers also analyzed brain tissue samples from ALS patients harboring FUS mutations and found that the amount of DNA damage in neurons in motor cortex was about double that found in normal brain tissue.
ALS patients with FUS mutations usually develop the disease early, before age 40. Only one of a person’s two copies of the FUS gene needs to be mutated to produce the disease. Tsai says that early in life, having one copy of the normal FUS gene may be enough to keep DNA repair going. “With aging, eventually the machinery is compromised and it contributes to neuronal demise,” she says.
The findings suggest that drugs that promote DNA damage repair, including activators of HDAC1 and SIRT1, could help combat the effects of ALS. SIRT1 activators are now being developed and have entered clinical trials to treat diabetes.
“There are numerous human inherited DNA-repair deficiency syndromes, many of which show neurodegeneration or other neurological defects. This new study now extends the spectrum of neuropathology caused by defects in DNA maintenance to include ALS,” says Peter McKinnon, a professor of genetics at St. Jude Children’s Research Hospital who was not part of the research team. “This study offers new avenues to explore in the quest for treatment strategies.”
Tsai’s lab is now studying whether there is a direct relationship between FUS and SIRT1. She also wants to determine whether the DNA damage that occurs in ALS patients after FUS is lost occurs in certain “hotspots” or is random. “I would speculate that there’s got to be hotspots in terms of where the DNA is damaged. But right now it remains speculation,” she says. “We really need to do the experiments and demonstrate whether that’s the case.”
The research was funded by the Simons Foundation, the National Institutes of Health, the Howard Hughes Medical Institute, the U.S. Department of Veteran Affairs, and the Muscular Dystrophy Association.