Penicillin and other antibiotics have revolutionized medicine, turning once-deadly diseases into easily treatable ailments. However, while antibiotics have been in use for more than 70 years, the exact mechanism by which they kill bacteria has remained a mystery.
Now a new study by MIT and Boston University researchers reveals the killing mechanism behind all three major classes of antibiotics: The drugs produce destructive molecules that fatally damage bacterial DNA through a long chain of cellular events.
Understanding the details of this mechanism could help scientists improve existing drugs, according to the researchers. Few new antibiotics have been developed in the past 40 years, and many strains of bacteria have become resistant to the drugs now available.
“One could enhance the killing efficacy of our current arsenal, reduce the required doses or resensitize strains to existing antibiotics,” says James Collins, a professor of biomedical engineering at BU, who collaborated with Graham Walker, MIT professor of biology, on a study appearing in the April 20 issue of Science.
Lead author of the paper is James Foti, a postdoc in Walker’s lab. Other authors are MIT postdoc Babho Devadoss and Jonathan Winkler, a recent PhD recipient in Collins’ lab.
Destructive radicals
In 2007, Collins showed that three classes of antibiotics — quinolones, beta-lactams and aminoglycosides — kill cells by producing highly destructive molecules known as hydroxyl radicals. At the time, he and others suspected that the radicals launch a general attack against any cell components they encounter.
“They react with almost everything,” Walker says. “They’ll go after lipids, they can oxidize proteins, they can oxidize DNA.” However, most of this damage is not fatal, the researchers found in the new study.
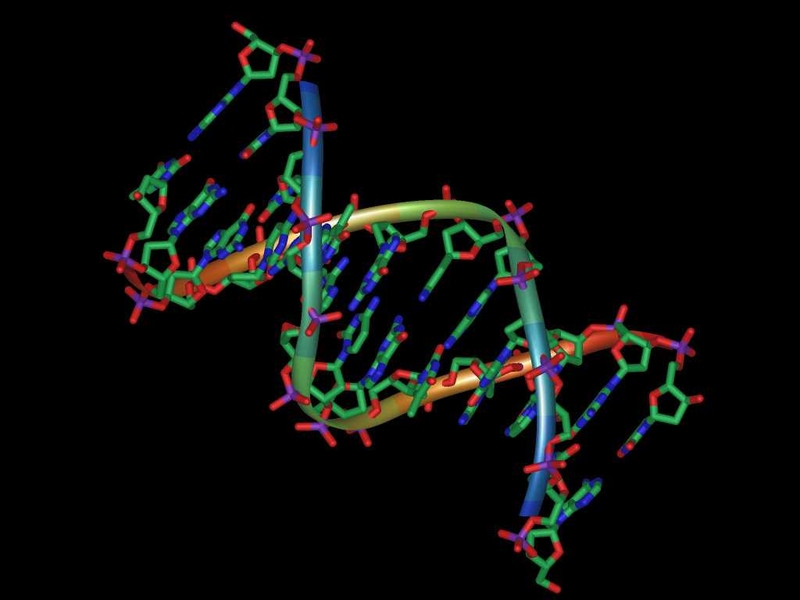
What proves deadly to bacteria is hydroxyl-induced damage to guanine, one of the four nucleotide bases that constitute DNA. When this damaged guanine is inserted into DNA, cells try to repair the damage but end up hastening their own death. This process “doesn’t account for all of the killing, but it accounts for a rather remarkable amount of it,” says Walker, who is an American Cancer Society Professor.
Walker’s studies of DNA repair enzymes led the researchers to suspect that this damaged guanine, known as oxidized guanine, might play a role in antibiotic-mediated cell death. In the first phase of their research, they showed that a specialized DNA-copying enzyme called DinB — part of a cell’s system for responding to DNA damage — is very good at utilizing the oxidized-guanine building block to synthesize DNA.
However, DinB not only inserts oxidized guanine opposite its correct base partner, cytosine, on the complementary strand when DNA is being copied, but also opposite its incorrect partner, adenine. The researchers found that, when too many oxidized guanines had been incorporated into new DNA strands, the cell’s unsuccessful efforts to remove these lesions resulted in death.
Based on these very basic DNA-repair studies, Walker and his colleagues hypothesized that the hydroxyl radicals produced by antibiotics might be setting off the same cascade of DNA damage. This turned out to be the case.
Once oxidized guanine caused by antibiotic treatment is inserted into DNA, a cellular system designed to repair DNA kicks into action. Specialized enzymes known as MutY and MutM make snips in the DNA to initiate repair processes that normally help the cells deal with the presence of oxidized guanine in their DNA. However, this repair is risky because it requires opening up the DNA double helix, severing one of its chains while the incorrect base is replaced. If two such repairs are undertaken in close proximity on opposite DNA strands, the DNA suffers a double-strand break, which is usually fatal to the cell.
“This system, which normally should be protecting you and keeping you very accurate, becomes your executioner,” Walker says.
Deborah Hung, a professor of microbiology and immunobiology at Harvard Medical School, says that the new study represents “the next important chapter as we’re going through a renaissance of understanding how antibiotics work. We used to think we knew, and now we’ve realized that all our simple assumptions were wrong, and it’s much more complex,” says Hung, who was not part of this study.
New targets
In some cases of antibiotic-induced DNA damage, the bacterial cell is able to save itself by repairing the double-strand break using a process called homologous recombination. Disabling the enzymes required for homologous recombination could increase bacteria’s sensitivity to antibiotics, the researchers say.
“Our work would suggest that proteins involved in repairing double-stranded DNA breaks could be very interesting targets to go after as a means to affect the killing efficacy of drugs,” Collins says.
The researchers, whose work was funded by the National Institutes of Health and Howard Hughes Medical Institute, also showed that an additional mechanism may be involved in cell deaths caused by one of the antibiotic classes, aminoglycosides: In cells treated with these antibiotics, oxidized guanine is incorporated into messenger RNA, resulting in incorrect proteins that, in turn, trigger more hydroxyl-radical production and hence more oxidized guanine. The researchers are now working to further advance their understanding of how antibiotics kill cells.
Now a new study by MIT and Boston University researchers reveals the killing mechanism behind all three major classes of antibiotics: The drugs produce destructive molecules that fatally damage bacterial DNA through a long chain of cellular events.
Understanding the details of this mechanism could help scientists improve existing drugs, according to the researchers. Few new antibiotics have been developed in the past 40 years, and many strains of bacteria have become resistant to the drugs now available.
“One could enhance the killing efficacy of our current arsenal, reduce the required doses or resensitize strains to existing antibiotics,” says James Collins, a professor of biomedical engineering at BU, who collaborated with Graham Walker, MIT professor of biology, on a study appearing in the April 20 issue of Science.
Lead author of the paper is James Foti, a postdoc in Walker’s lab. Other authors are MIT postdoc Babho Devadoss and Jonathan Winkler, a recent PhD recipient in Collins’ lab.
Destructive radicals
In 2007, Collins showed that three classes of antibiotics — quinolones, beta-lactams and aminoglycosides — kill cells by producing highly destructive molecules known as hydroxyl radicals. At the time, he and others suspected that the radicals launch a general attack against any cell components they encounter.
“They react with almost everything,” Walker says. “They’ll go after lipids, they can oxidize proteins, they can oxidize DNA.” However, most of this damage is not fatal, the researchers found in the new study.
What proves deadly to bacteria is hydroxyl-induced damage to guanine, one of the four nucleotide bases that constitute DNA. When this damaged guanine is inserted into DNA, cells try to repair the damage but end up hastening their own death. This process “doesn’t account for all of the killing, but it accounts for a rather remarkable amount of it,” says Walker, who is an American Cancer Society Professor.
Walker’s studies of DNA repair enzymes led the researchers to suspect that this damaged guanine, known as oxidized guanine, might play a role in antibiotic-mediated cell death. In the first phase of their research, they showed that a specialized DNA-copying enzyme called DinB — part of a cell’s system for responding to DNA damage — is very good at utilizing the oxidized-guanine building block to synthesize DNA.
However, DinB not only inserts oxidized guanine opposite its correct base partner, cytosine, on the complementary strand when DNA is being copied, but also opposite its incorrect partner, adenine. The researchers found that, when too many oxidized guanines had been incorporated into new DNA strands, the cell’s unsuccessful efforts to remove these lesions resulted in death.
Based on these very basic DNA-repair studies, Walker and his colleagues hypothesized that the hydroxyl radicals produced by antibiotics might be setting off the same cascade of DNA damage. This turned out to be the case.
Once oxidized guanine caused by antibiotic treatment is inserted into DNA, a cellular system designed to repair DNA kicks into action. Specialized enzymes known as MutY and MutM make snips in the DNA to initiate repair processes that normally help the cells deal with the presence of oxidized guanine in their DNA. However, this repair is risky because it requires opening up the DNA double helix, severing one of its chains while the incorrect base is replaced. If two such repairs are undertaken in close proximity on opposite DNA strands, the DNA suffers a double-strand break, which is usually fatal to the cell.
“This system, which normally should be protecting you and keeping you very accurate, becomes your executioner,” Walker says.
Deborah Hung, a professor of microbiology and immunobiology at Harvard Medical School, says that the new study represents “the next important chapter as we’re going through a renaissance of understanding how antibiotics work. We used to think we knew, and now we’ve realized that all our simple assumptions were wrong, and it’s much more complex,” says Hung, who was not part of this study.
New targets
In some cases of antibiotic-induced DNA damage, the bacterial cell is able to save itself by repairing the double-strand break using a process called homologous recombination. Disabling the enzymes required for homologous recombination could increase bacteria’s sensitivity to antibiotics, the researchers say.
“Our work would suggest that proteins involved in repairing double-stranded DNA breaks could be very interesting targets to go after as a means to affect the killing efficacy of drugs,” Collins says.
The researchers, whose work was funded by the National Institutes of Health and Howard Hughes Medical Institute, also showed that an additional mechanism may be involved in cell deaths caused by one of the antibiotic classes, aminoglycosides: In cells treated with these antibiotics, oxidized guanine is incorporated into messenger RNA, resulting in incorrect proteins that, in turn, trigger more hydroxyl-radical production and hence more oxidized guanine. The researchers are now working to further advance their understanding of how antibiotics kill cells.