Prion diseases, such as mad cow disease in cattle and Creutzfeldt-Jakob disease in humans, have stumped scientists for decades with a complex "whodunit" complete with many suspects and a missing murder weapon. New research from Susan Lindquist, director of the Whitehead Institute for Biomedical Research and MIT professor of biology, and Jiyan Ma of Ohio State University suggests a unifying theory that can help explain how these devastating diseases get started and how they kill.
Unlike other infectious diseases that are linked to pathogens such as bacteria and viruses, these diseases have a unique and mysterious connection to a misfolded protein. The brain of a victim killed by prion diseases is typically clogged with clumps of prion protein (PrP) in a rare, misfolded state called PrPSc. Controversy has long raged about how these diseases get started and if the clumps of PrPSc actually kill brain cells - in some forms of the disease, clumps aren't even there. The culprit responsible for the death of neurons is still a mystery.
Lindquist and Ma's results, published in two papers in the Oct. 17 online issue of Science, establish a mechanism for how normal PrP can convert into a highly toxic form that kills neurons and is distinct from the self-propagating PrPSc form of the protein.
"This is an entirely new view of the toxic species," said Lindquist. "We think this toxicity wasn't realized before, simply because such small amounts of PrP are required for cell death and other forms of the protein are present at much higher concentrations." Mammalian Prion Proteins
Mad cow disease came to the attention of scientists in 1986 when an epidemic of a new neurological disease erupted in British cattle herds. Almost 10 years later, fears accelerated when young people, mostly in Britain, contracted the same disease, apparently from eating infected meat. (In people, the disease was previously found only in the elderly.) To date, approximately 150 young people have contracted this new variant of Creutzfeldt-Jakob disease. While scientists hope this number will remain small, it's hard to tell since the incubation period can be as long as 20 years. Recently, a related disease, known as "wasting disease," has been spreading across the United States in deer and elk populations.
All mammals, including humans, make the PrP prion protein, but the rare, misfolded PrPSc state is found only in mammals with transmissible prion diseases. Remarkably, this shape change appears to be contagious, much like one bad apple ruining the bunch. Once PrPSc forms, it can coax other prion proteins into this alternative folding pattern and cause them to clump together. PrPSc is widely believed to be the infectious agent in transmissible forms of the disease, responsible for starting the deadly chain of PrP misfolding events. But the mechanism for triggering the initial, rare PrPSc conformation was unknown.
The very idea of prions makes many scientists uneasy because it violates the fundamental principle that the presence of nucleic acids, such as DNA, is essential both to inheritance and infection. Yet prions produce clumps in the brain that are infectious even though no nucleic acid seems to be involved. It's All in the Fold
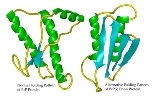
Like simple sheets of paper that fold into origami figures, proteins fold into a vast array of complex and beautiful shapes. Sometimes a protein folds incorrectly by chance; at other times a mutation in the protein makes it more likely to misfold. Because misfolded proteins can cause a cell to malfunction, the cell uses a stringent quality control system to eliminate them.
Misfolded PrP, for instance, doesn't get transported to the surface of the cell, its normal destination. Instead, when the cell detects misfolded PrP, it's spit into the cellular space called the cytosol. There, protein-degrading machines called proteasomes act as garbage disposals to eliminate misfolded proteins. Usually, PrP is degraded so rapidly that it is undetectable. However, when proteasome activity is compromised, as might naturally occur with stress and aging, PrP accumulates in the cytosol.
The Lindquist lab used proteasome inhibitors to block the cell's garbage disposal to see what would happen when misfolded PrP accumulated in the cytosol. Surprisingly, neurons died even before PrPSc clumps formed.
Depending on how fast PrP accumulated in the cytosol, it sometimes converted to a PrPSc-like conformation. "When proteins are delivered to the cytosol for destruction, they're unfolded," Lindquist explained. "We think conversion occurs when these wobbly, sticky PrP molecules find others in the same state. If enough of them get together at the same time, they can acquire this new structure."
In the lab, once this new conformation appeared, it recruited other PrP molecules to make the same shape change. The shape changes kept occurring even after the inhibitor was removed and the degradation of other proteins resumed. PrPSc, however, did not seem to be killing the neurons. That is, the fraction of PrP that converts to the PrPSc-like form did not determine cell death.
This work demonstrated the first natural, biological mechanism for producing the PrPSc conformation from scratch inside the cell. It also shows that the self-perpetuating structural change is an intrinsic - and unusual - property of the PrP protein itself.
"That's a very important finding," Lindquist said. "But it doesn't prove that conversion is sufficient for transmission from animal to animal. That has yet to be tested, and those experiments can take a long time."
Next, to avoid artifacts that might be caused by the inhibitors, the researchers simply made transgenic mice that would make the normal PrP protein directly in the cytosol. It was deadly. Even very small amounts of soluble PrP caused massive degeneration of neurons, quickly destroying the brain. Mice suffered from loss of muscle control, similar to patients with Creutzfeldt-Jakob disease and other prion diseases.
In fact, the toxicity of cytosolic PrP is so great that it may have evolved as a way to kill neurons with PrP folding problems. It would be better for an individual neuron with folding problems to die rather than to risk the possibility that PrP might accumulate at sufficient levels to produce the infectious PrPSc form, which could then spread to other cells and kill the whole brain.
With a better understanding of how prion diseases initiate and progress, scientists will be better able to design therapeutic approaches for treatment.
A version of this article appeared in MIT Tech Talk on October 30, 2002.