Learning, memory, and behavioral disorders can arise when the connections between neurons, called synapses, do not change properly in response to experience. Scientists have studied this “synaptic plasticity” for decades, but a new study by researchers at MIT’s Picower Institute for Learning and Memory highlights several surprises about some of the basic mechanisms by which it happens. Getting to the bottom of what underlies some of those surprises, the research further suggests, could yield new treatments for a disorder called Fragile X that causes autism.
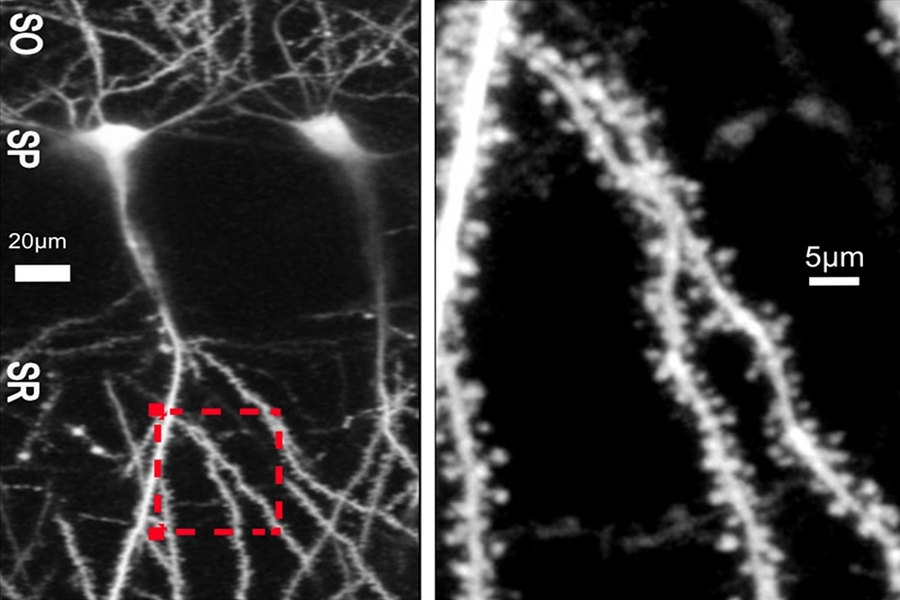
Two classic forms of synaptic plasticity are that synapses either get stronger or weaker and that the tiny spine structures that support them get bigger or smaller. For a long time, the field’s working assumption has been that these functional and structural changes were closely associated: Strengthening went along with an increase in spine size and weakening preceded spine shrinkage. But the study published in Molecular Psychiatry adds specific evidence to support a more recent view, backed by other recent studies, that those correlations are not always true.
“We saw these breakdowns of correlation between structure and function,” says Mark Bear, Picower Professor in the Department of Brain and Cognitive Sciences and senior author of the study. “One conclusion is you can’t use spine size as a proxy for synaptic strength — you can have weak synapses with big bulbous spines. We are not the only ones to make this case, but the new results in this study are very clear.”
The study’s co-lead authors are former lab members Aurore Thomazeau and Miquel Bosch.
To conduct the study, the team stimulated plasticity via two different neural receptors (called mGluR5 and NMDAR) under two different conditions (neurotypical rodents and ones engineered with the mutation that causes Fragile X). In Fragile X, Bear’s lab has found the lack of the protein FMRP leads to excess synthesis of other proteins that cause synapses to weaken too much in a brain region called the hippocampus, which is a crucial area for memory formation.
The first surprise of the study was that activating mGluR5 receptors induced the weakening, called long-term depression (LTD), but did not lead to any spine shrinkage in either Fragile X or control mice for at least an hour. In other words, the structural change that was assumed to go along with the functional change didn’t actually occur.
In the NMDAR case, the two forms of plasticity did occur together, both in control and Fragile X rodents, but not without a few more surprises lurking just beneath the surface that further dissociated functional and structural plasticity. When the team blocked a flow of ions (and therefore electric current) in the NMDAR synapses, that only prevented the weakening, not the shrinking. To prevent the shrinking in control rodents, the researchers had to do something different: inhibit protein synthesis either directly or by inhibiting a regulatory protein called mTORC1.
“It was quite amazing to us,” Bear says. “We are following up on that aggressively to better understand that signaling.”
If several of the surprises in the study are disruptive, Bear said, another one may provide new hope for treating Fragile X. That’s because while Bear’s lab has focused on intervening in the mGluR pathway to treat Fragile X, the new experiments involving NMDAR may reveal an additional avenue.
When the team tried to prevent spines from shrinking via NMDAR in Fragile X rodents by inhibiting protein synthesis or mTORC1 (like they did in the controls), they found it didn’t work. It was as if there was already too much of some protein that promotes the shrinkage. The team was even able to replicate this Fragile X phenomenon in the controls by first stimulating mGluR5 — and an ensuing excess of protein synthesis — and then following up with the NMDAR activation.
As a nod to both the mystery and the disorder, Bear has begun to refer to this conjectured potential shrinkage-promoting molecule as “protein X.”
“The question is what is protein X,” Bear says. “The evidence is quite strong that there is a rapidly turned over protein X that is wreaking havoc in Fragile X. Now the hunt is on. We’ll be really excited to find it.”
In addition to Thomazeau, Bosch and Bear, the paper’s other authors are Sofia Essayan-Perez, Stephanie Barnes, and Héctor de Jesús-Cortés.
A Beatriu de Pinos fellowship, the FRAXA Foundation, a Marie Curie Reintegration Grant, the U.S. National Institutes of Health and the JPB Foundation supported the research.