Williams Syndrome, a rare neurodevelopmental disorder that affects about 1 in 10,000 babies born in the United States, produces a range of symptoms including cognitive impairments, cardiovascular problems, and extreme friendliness, or hypersociability.
In a study of mice, MIT neuroscientists have garnered new insight into the molecular mechanisms that underlie this hypersociability. They found that loss of one of the genes linked to Williams Syndrome leads to a thinning of the fatty layer that insulates neurons and helps them conduct electrical signals in the brain.
The researchers also showed that they could reverse the symptoms by boosting production of this coating, known as myelin. This is significant, because while Williams Syndrome is rare, many other neurodevelopmental disorders and neurological conditions have been linked to myelination deficits, says Guoping Feng, the James W. and Patricia Poitras Professor of Neuroscience and a member of MIT’s McGovern Institute for Brain Research.
“The importance is not only for Williams Syndrome,” says Feng, who is one of the senior authors of the study. “In other neurodevelopmental disorders, especially in some of the autism spectrum disorders, this could be potentially a new direction to look into, not only the pathology but also potential treatments.”
Zhigang He, a professor of neurology and ophthalmology at Harvard Medical School, is also a senior author of the paper, which appears in the April 22 issue of Nature Neuroscience. Former MIT postdoc Boaz Barak, currently a principal investigator at Tel Aviv University in Israel, is the lead author and a senior author of the paper.
Impaired myelination
Williams Syndrome, which is caused by the loss of one of the two copies of a segment of chromosome 7, can produce learning impairments, especially for tasks that require visual and motor skills, such as solving a jigsaw puzzle. Some people with the disorder also exhibit poor concentration and hyperactivity, and they are more likely to experience phobias.
In this study, the researchers decided to focus on one of the 25 genes in that segment, known as Gtf2i. Based on studies of patients with a smaller subset of the genes deleted, scientists have linked the Gtf2i gene to the hypersociability seen in Williams Syndrome.
Working with a mouse model, the researchers devised a way to knock out the gene specifically from excitatory neurons in the forebrain, which includes the cortex, the hippocampus, and the amygdala (a region important for processing emotions). They found that these mice did show increased levels of social behavior, measured by how much time they spent interacting with other mice. The mice also showed deficits in fine motor skills and increased nonsocial related anxiety, which are also symptoms of Williams Syndrome.
Next, the researchers sequenced the messenger RNA from the cortex of the mice to see which genes were affected by loss of Gtf2i. Gtf2i encodes a transcription factor, so it controls the expression of many other genes. The researchers found that about 70 percent of the genes with significantly reduced expression levels were involved in the process of myelination.
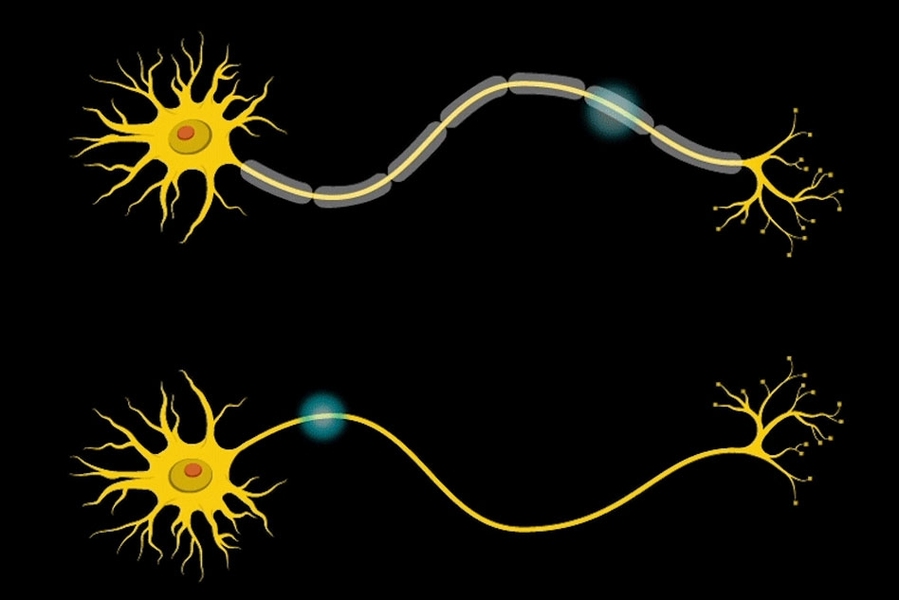
“Myelin is the insulation layer that wraps the axons that extend from the cell bodies of neurons,” Barak says. “When they don’t have the right properties, it will lead to faster or slower electrical signal transduction, which affects the synchronicity of brain activity.”
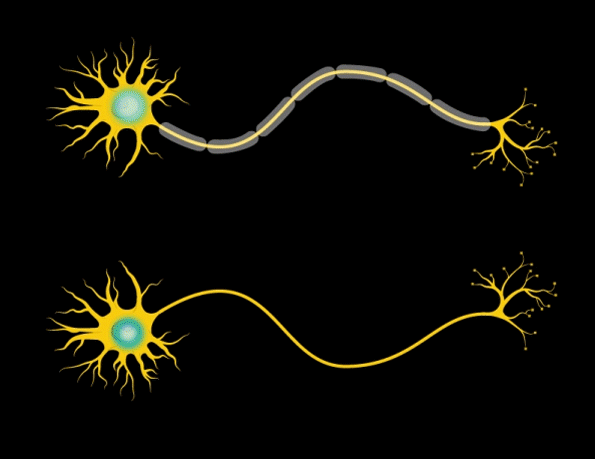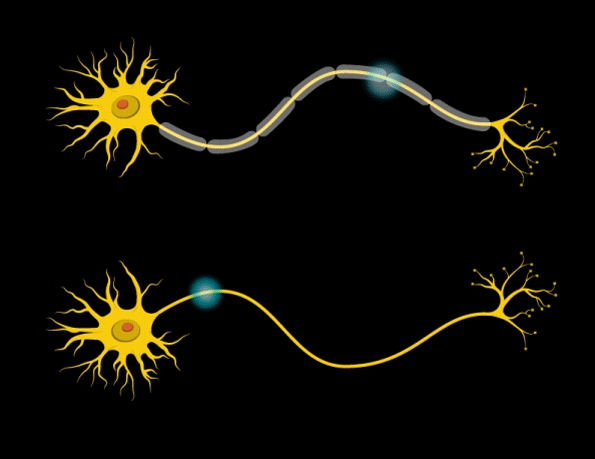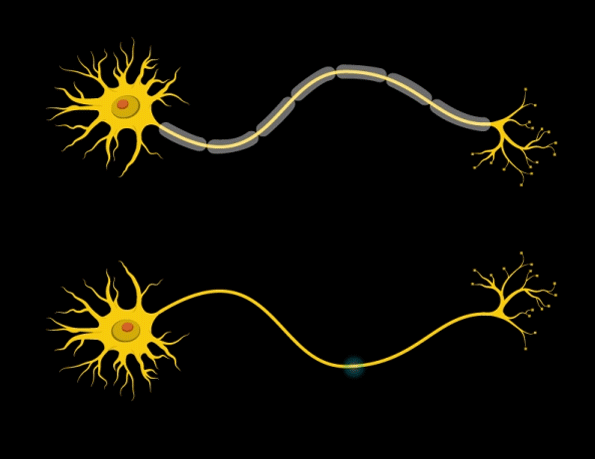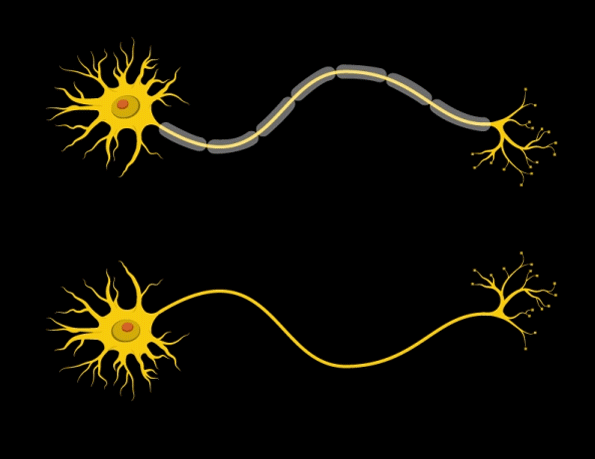
Further studies revealed that the mice had only about half the normal number of mature oligodendrocytes — the brain cells that produce myelin. However, the number of oligodendrocyte precursor cells was normal, so the researchers suspect that the maturation and differentiation processes of these cells are somehow impaired when Gtf2i is missing in the neurons.
This was surprising because Gtf2i was not knocked out in oligodendrocytes or their precursors. Thus, knocking out the gene in neurons may somehow influence the maturation process of oligodendrocytes, the researchers suggest. It is still unknown how this interaction might work.
“That’s a question we are interested in, but we don’t know whether it’s a secreted factor, or another kind of signal or activity,” Feng says.
In addition, the researchers found that the myelin surrounding axons of the forebrain was significantly thinner than in normal mice. Furthermore, electrical signals were smaller, and took more time to cross the brain in mice with Gtf2i missing.
The study is an example of pioneering research into the contribution of glial cells, which include oligodendrocytes, to neuropsychiatric disorders, says Doug Fields, chief of the nervous system development and plasticity section of the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
“Traditionally myelin was only considered in the context of diseases that destroy myelin, such as multiple sclerosis, which prevents transmission of neural impulses. More recently it has become apparent that more subtle defects in myelin can impair neural circuit function, by causing delays in communication between neurons,” says Fields, who was not involved in the research.
Symptom reversal
It remains to be discovered precisely how this reduction in myelination leads to hypersociability. The researchers suspect that the lack of myelin affects brain circuits that normally inhibit social behaviors, making the mice more eager to interact with others.
“That’s probably the explanation, but exactly which circuits and how does it work, we still don’t know,” Feng says.
The researchers also found that they could reverse the symptoms by treating the mice with drugs that improve myelination. One of these drugs, an FDA-approved antihistamine called clemastine fumarate, is now in clinical trials to treat multiple sclerosis, which affects myelination of neurons in the brain and spinal cord. The researchers believe it would be worthwhile to test these drugs in Williams Syndrome patients because they found thinner myelin and reduced numbers of mature oligodendrocytes in brain samples from human subjects who had Williams Syndrome, compared to typical human brain samples.
“Mice are not humans, but the pathology is similar in this case, which means this could be translatable,” Feng says. “It could be that in these patients, if you improve their myelination early on, it could at least improve some of the conditions. That’s our hope.”
Such drugs would likely help mainly the social and fine-motor issues caused by Williams Syndrome, not the symptoms that are produced by deletion of other genes, the researchers say. They may also help treat other disorders, such as autism spectrum disorders, in which myelination is impaired in some cases, Feng says.
“We think this can be expanded into autism and other neurodevelopmental disorders. For these conditions, improved myelination may be a major factor in treatment,” he says. “We are now checking other animal models of neurodevelopmental disorders to see whether they have myelination defects, and whether improved myelination can improve some of the pathology of the defects.”
The research was funded by the Simons Foundation, the Poitras Center for Affective Disorders Research at MIT, the Stanley Center for Psychiatric Research at the Broad Institute of MIT and Harvard, and the Simons Center for the Social Brain at MIT.








