In recent years, machine learning has been proving a valuable tool for identifying new materials with properties optimized for specific applications. Working with large, well-defined data sets, computers learn to perform an analytical task to generate a correct answer and then use the same technique on an unknown data set.
While that approach has guided the development of valuable new materials, they’ve primarily been organic compounds, notes Heather Kulik PhD ’09, an assistant professor of chemical engineering. Kulik focuses instead on inorganic compounds — in particular, those based on transition metals, a family of elements (including iron and copper) that have unique and useful properties. In those compounds — known as transition metal complexes — the metal atom occurs at the center with chemically bound arms, or ligands, made of carbon, hydrogen, nitrogen, or oxygen atoms radiating outward.
Transition metal complexes already play important roles in areas ranging from energy storage to catalysis for manufacturing fine chemicals — for example, for pharmaceuticals. But Kulik thinks that machine learning could further expand their use. Indeed, her group has been working not only to apply machine learning to inorganics — a novel and challenging undertaking — but also to use the technique to explore new territory. “We were interested in understanding how far we could push our models to do discovery — to make predictions on compounds that haven’t been seen before,” says Kulik.
Sensors and computers
For the past four years, Kulik and Jon Paul Janet, a graduate student in chemical engineering, have been focusing on transition metal complexes with “spin” — a quantum mechanical property of electrons. Usually, electrons occur in pairs, one with spin up and the other with spin down, so they cancel each other out and there’s no net spin. But in a transition metal, electrons can be unpaired, and the resulting net spin is the property that makes inorganic complexes of interest, says Kulik. “Tailoring how unpaired the electrons are gives us a unique knob for tailoring properties.”
A given complex has a preferred spin state. But add some energy — say, from light or heat — and it can flip to the other state. In the process, it can exhibit changes in macroscale properties such as size or color. When the energy needed to cause the flip — called the spin-splitting energy — is near zero, the complex is a good candidate for use as a sensor, or perhaps as a fundamental component in a quantum computer.
Chemists know of many metal-ligand combinations with spin-splitting energies near zero, making them potential “spin-crossover” (SCO) complexes for such practical applications. But the full set of possibilities is vast. The spin-splitting energy of a transition metal complex is determined by what ligands are combined with a given metal, and there are almost endless ligands from which to choose. The challenge is to find novel combinations with the desired property to become SCOs — without resorting to millions of trial-and-error tests in a lab.
Translating molecules into numbers
The standard way to analyze the electronic structure of molecules is using a computational modeling method called density functional theory, or DFT. The results of a DFT calculation are fairly accurate — especially for organic systems — but performing a calculation for a single compound can take hours, or even days. In contrast, a machine learning tool called an artificial neural network (ANN) can be trained to perform the same analysis and then do it in just seconds. As a result, ANNs are much more practical for looking for possible SCOs in the huge space of feasible complexes.
Because an ANN requires a numerical input to operate, the researchers’ first challenge was to find a way to represent a given transition metal complex as a series of numbers, each describing a selected property. There are rules for defining representations for organic molecules, where the physical structure of a molecule tells a lot about its properties and behavior. But when the researchers followed those rules for transition metal complexes, it didn’t work. “The metal-organic bond is very tricky to get right,” says Kulik. “There are unique properties of the bonding that are more variable. There are many more ways the electrons can choose to form a bond.” So the researchers needed to make up new rules for defining a representation that would be predictive in inorganic chemistry.
Using machine learning, they explored various ways of representing a transition metal complex for analyzing spin-splitting energy. The results were best when the representation gave the most emphasis to the properties of the metal center and the metal-ligand connection and less emphasis to the properties of ligands farther out. Interestingly, their studies showed that representations that gave more equal emphasis overall worked best when the goal was to predict other properties, such as the ligand-metal bond length or the tendency to accept electrons.
Testing the ANN
As a test of their approach, Kulik and Janet — assisted by Lydia Chan, a summer intern from Troy High School in Fullerton, California — defined a set of transition metal complexes based on four transition metals — chromium, manganese, iron, and cobalt — in two oxidation states with 16 ligands (each molecule can have up to two). By combining those building blocks, they created a “search space” of 5,600 complexes — some of them familiar and well-studied, and some of them totally unknown.
In previous work, the researchers had trained an ANN on thousands of compounds that were well-known in transition metal chemistry. To test the trained ANN’s ability to explore a new chemical space to find compounds with the targeted properties, they tried applying it to the pool of 5,600 complexes, 113 of which it had seen in the previous study.
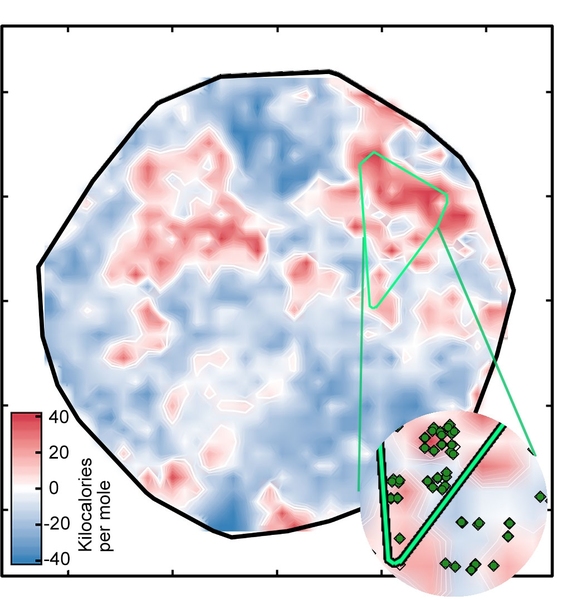
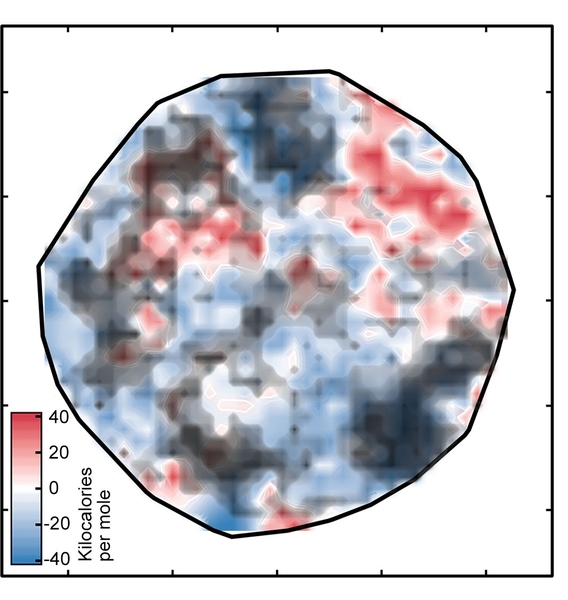
The result was the plot labeled "Figure 1" in the slideshow above, which sorts the complexes onto a surface as determined by the ANN. The white regions indicate complexes with spin-splitting energies within 5 kilo-calories per mole of zero, meaning that they are potentially good SCO candidates. The red and blue regions represent complexes with spin-splitting energies too large to be useful. The green diamonds that appear in the inset show complexes that have iron centers and similar ligands — in other words, related compounds whose spin-crossover energies should be similar. Their appearance in the same region of the plot is evidence of the good correspondence between the researchers’ representation and key properties of the complex.
But there’s one catch: Not all of the spin-splitting predictions are accurate. If a complex is very different from those on which the network was trained, the ANN analysis may not be reliable — a standard problem when applying machine learning models to discovery in materials science or chemistry, notes Kulik. Using an approach that looked successful in their previous work, the researchers compared the numeric representations for the training and test complexes and ruled out all the test complexes where the difference was too great.
Focusing on the best options
Performing the ANN analysis of all 5,600 complexes took just an hour. But in the real world, the number of complexes to be explored could be thousands of times larger — and any promising candidates would require a full DFT calculation. The researchers therefore needed a method of evaluating a big data set to identify any unacceptable candidates even before the ANN analysis. To that end, they developed a genetic algorithm — an approach inspired by natural selection — to score individual complexes and discard those deemed to be unfit.
To prescreen a data set, the genetic algorithm first randomly selects 20 samples from the full set of complexes. It then assigns a “fitness” score to each sample based on three measures. First, is its spin-crossover energy low enough for it to be a good SCO? To find out, the neural network evaluates each of the 20 complexes. Second, is the complex too far away from the training data? If so, the spin-crossover energy from the ANN may be inaccurate. And finally, is the complex too close to the training data? If so, the researchers have already run a DFT calculation on a similar molecule, so the candidate is not of interest in the quest for new options.
Based on its three-part evaluation of the first 20 candidates, the genetic algorithm throws out unfit options and saves the fittest for the next round. To ensure the diversity of the saved compounds, the algorithm calls for some of them to mutate a bit. One complex may be assigned a new, randomly selected ligand, or two promising complexes may swap ligands. After all, if a complex looks good, then something very similar could be even better — and the goal here is to find novel candidates. The genetic algorithm then adds some new, randomly chosen complexes to fill out the second group of 20 and performs its next analysis. By repeating this process a total of 21 times, it produces 21 generations of options. It thus proceeds through the search space, allowing the fittest candidates to survive and reproduce, and the unfit to die out.
Performing the 21-generation analysis on the full 5,600-complex data set required just over five minutes on a standard desktop computer, and it yielded 372 leads with a good combination of high diversity and acceptable confidence. The researchers then used DFT to examine 56 complexes randomly chosen from among those leads, and the results confirmed that two-thirds of them could be good SCOs.
While a success rate of two-thirds may not sound great, the researchers make two points. First, their definition of what might make a good SCO was very restrictive: For a complex to survive, its spin-splitting energy had to be extremely small. And second, given a space of 5,600 complexes and nothing to go on, how many DFT analyses would be required to find 37 leads? As Janet notes, “It doesn’t matter how many we evaluated with the neural network because it’s so cheap. It’s the DFT calculations that take time.”
Best of all, using their approach enabled the researchers to find some unconventional SCO candidates that wouldn’t have been thought of based on what’s been studied in the past. “There are rules that people have — heuristics in their heads — for how they would build a spin-crossover complex,” says Kulik. “We showed that you can find unexpected combinations of metals and ligands that aren’t normally studied but can be promising as spin-crossover candidates.”
Sharing the new tools
To support the worldwide search for new materials, the researchers have incorporated the genetic algorithm and ANN into "molSimplify," the group’s online, open-source software toolkit that anyone can download and use to build and simulate transition metal complexes. To help potential users, the site provides tutorials that demonstrate how to use key features of the open-source software codes. Development of molSimplify began with funding from the MIT Energy Initiative in 2014, and all the students in Kulik’s group have contributed to it since then.
The researchers continue to improve their neural network for investigating potential SCOs and to post updated versions of molSimplify. Meanwhile, others in Kulik’s lab are developing tools that can identify promising compounds for other applications. For example, one important area of focus is catalyst design. Graduate student in chemistry Aditya Nandy is focusing on finding a better catalyst for converting methane gas to an easier-to-handle liquid fuel such as methanol — a particularly challenging problem. “Now we have an outside molecule coming in, and our complex — the catalyst — has to act on that molecule to perform a chemical transformation that takes place in a whole series of steps,” says Nandy. “Machine learning will be super-useful in figuring out the important design parameters for a transition metal complex that will make each step in that process energetically favorable.”
This research was supported by the U.S. Department of the Navy’s Office of Naval Research, the U.S. Department of Energy, the National Science Foundation, and the MIT Energy Initiative Seed Fund Program. Jon Paul Janet was supported in part by an MIT-Singapore University of Technology and Design Graduate Fellowship. Heather Kulik has received a National Science CAREER Award (2019) and an Office of Naval Research Young Investigator Award (2018), among others.
This article appears in the Spring 2019 issue of Energy Futures, the magazine of the MIT Energy Initiative.