If there’s one fact that most people retain from elementary biology, it’s that mitochondria are the powerhouses of the cell. As such, they break down molecules and manufacture new ones to generate the fuel necessary for life. But mitochondria rely on a stream of proteins to sustain this energy production. Nearly all their proteins are manufactured in the surrounding gel-like cytoplasm, and must be imported into the mitochondria to keep the powerhouse running.
A duo of MIT biologists has revealed what happens when a traffic jam of proteins at the surface of the mitochondria prevents proper import. They describe how the mitochondria communicate with the rest of the cell to signal a problem, and how the cell responds to protect the mitochondria. This newly-discovered molecular pathway, called mitoCPR, detects import mishaps and preserves mitochondrial function in the midst of such stress.
“This is the first mechanism identified that surveils mitochondrial protein import, and helps mitochondria when they can't get the proteins they need,” says Angelika Amon, the Kathleen and Curtis Marble Professor of Cancer Research in the MIT Department of Biology, who is also a member of the Koch Institute for Integrative Cancer Research at MIT, a Howard Hughes Medical Institute Investigator, and senior author of the study. “Responses to mitochondrial stress have been established before, but this one specifically targets the surface of the mitochondria, clearing out the misfolded proteins that are stuck in the pores.”
Hilla Weidberg, a postdoc in Amon’s lab, is the lead author of the study, which appears in Science on April 13.
Fueling the powerhouse
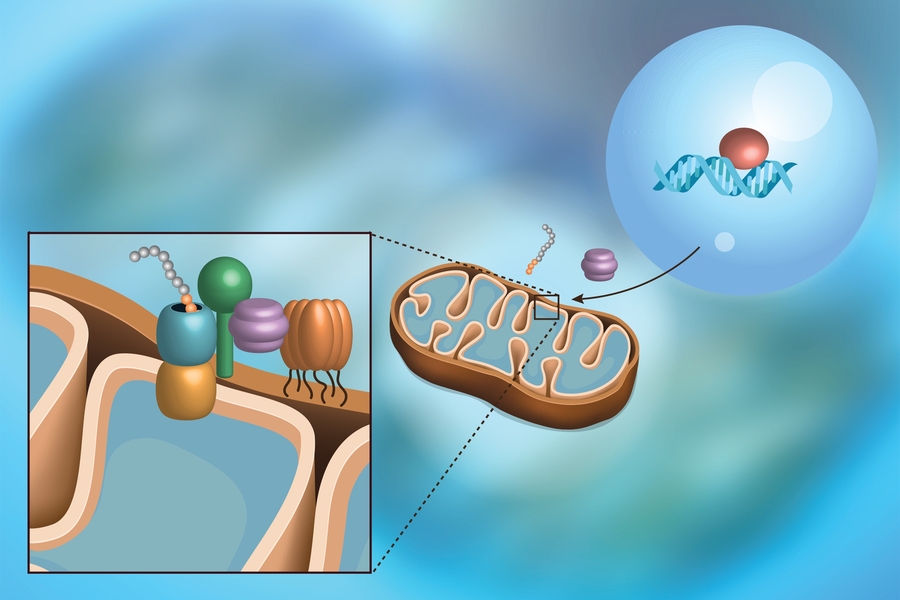
Mitochondria likely began as independent entities long ago, before being engulfed by host cells. They eventually gave up control and moved most of their important genes to a different organelle, the nucleus, where the rest of the cell’s genetic blueprint is stored. The protein products from these genes are ultimately made in the cytoplasm outside the nucleus, and then guided to the mitochondria. These “precursor” proteins contain a special molecular zip code that guides them through the channels at the surface of the mitochondria to their respective homes.
The proteins must be unfolded and delicately threaded through the narrow channels in order to enter the mitochondria. This creates a precarious situation; if the demand is too high, or the proteins are folded when they shouldn’t be, a bottleneck forms that none shall pass. This can simply occur when the mitochondria expand to make more of themselves, or in diseases like deafness-dystonia syndrome and Huntington’s.
“The machinery that we’ve identified seems to evict proteins that are sitting on the surface of the mitochondria and sends them for degradation,” Amon says. “Another possibility is that this mitoCPR pathway might actually unfold these proteins, and in doing so give them a second chance to be pushed through the membrane.”
Two other pathways were recently identified in yeast that also respond to accumulated mitochondrial proteins. However, both simply clear protein refuse from the cytoplasm around the mitochondria, rather than removing the proteins collecting on the mitochondria themselves.
“We knew about various responses to mitochondrial stress, but no one had described a response to protein import defects that specifically protected the mitochondria, and that’s exactly what mitoCPR does,” Weidberg says. “We wanted to know how the cell reacts to these problems, so we set out to overload the import machinery, causing many proteins to rush into the mitochondrion at the same time and clog the pores, triggering a cellular response.”
“What makes our cells absolutely dependent on mitochondria is one of those million-dollar questions in cell biology,” says Vlad Denic, professor of molecular and cellular biology at Harvard University. “This study reveals an interesting flip-side to that question: When you make mitochondrial life artificially tough, are they programmed to say ‘help us’ so the host cell comes to their rescue? The possible ramifications of such work in terms of human development and disease could be very impressive.”
A pathway to understanding
Roughly two decades ago, researchers began to notice that the genes required to defend cells against drugs and other foreign substances — together, called the multidrug resistance (MDR) response — were also expressed in yeast mitochondrial mutants for some unknown reason. This suggested that the protein in charge of binding to the DNA and initiating the MDR response must have a dual purpose, sometimes triggering a second, separate pathway as well. But precisely how this second pathway related to mitochondria remained a mystery.
“Twenty years ago, scientists recognized mitoCPR as some kind of mechanism against mitochondrial dysfunction,” Weidberg says. “Today we’ve finally characterized it, given it a name, and identified its precise function: to help mitochondrial protein import.”
As the import process slows, Amon and Weidberg determined that the protein that initiates mitoCPR — the transcription factor Pdr3 — binds to DNA within the nucleus, inducing the expression of a gene known as CIS1. The resultant Cis1 protein binds to the channel at the surface of the mitochondrion, and recruits yet another protein, the AAA+ adenosine triphosphatase Msp1, to help clear unimported proteins from the mitochondrial surface and mediate their degradation. Although the MDR response pathway differs from that of mitoCPR, both rely on Pdr3 activation. In fact, mitoCPR requires it.
“Whether the two pathways interact with one another is a very interesting question,” Amon says. “The mitochondria make a lot of biosynthetic molecules, and blocking that function by messing with protein import could lead to the accumulation of intermediate metabolites. These can be toxic to the cell, so you could imagine that activating the MDR response might pump out harmful intermediates.”
The question of what activates Pdr3 to initiate mitoCPR is still unclear, but Weidberg has some ideas related to signals stemming from the build-up of toxic metabolite intermediates. It’s also yet to be determined whether an analogous pathway exists in more complex organisms, although there is some evidence that the mitochondria do communicate with the nucleus in other eukaryotes besides yeast.
“This was just such a classic study,” Amon says. “There were no sophisticated high-throughput methodologies, just traditional, simple molecular biology and cell biology assays with a few microscopes. It’s almost like something you’d see out of the 1980s. But that just goes to show — to this day — that’s how many discoveries are made.”
The research was funded by the National Institutes of Health and by the Koch Institute Support (core) Grant from the National Cancer Institute. Amon is also an investigator of the Howard Hughes Medical Institute and the Glenn Foundation for Biomedical Research. Weidberg was supported by the Jane Coffin Childs Memorial Fund, the European Molecular Biology Organization Long-Term Fellowship, and the Israel National Postdoctoral Program for Advancing Women in Science.