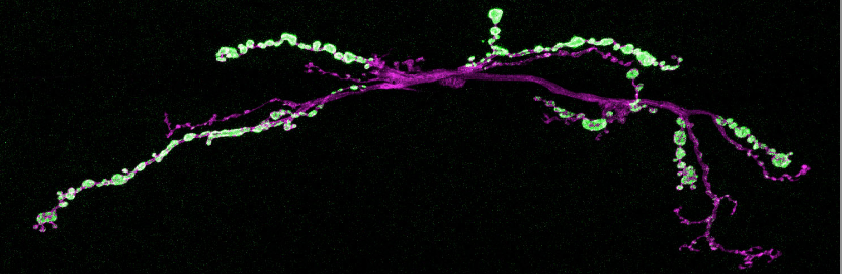
Throughout the animal kingdom, cells encapsulate molecules and proteins — that they move within or between — in tiny vesicles, which release their contents when they fuse with another membrane. Vesicles also package the chemical signals, or neurotransmitters, that leap from neuron to neuron in the brain’s communication network, but neurons more tightly control the release of these signals. In schizophrenia, Parkinson’s disease and other neurological disorders, however, this control breaks down, which may contribute to deficits in information processing. And researchers are seeking an explanation for the loss of the normal control mechanism.
Two new MIT studies now demonstrate how neurons have adapted the cell’s standard fusion machinery to regulate the release of neurotransmitters at the neuron’s chemical junctions called synapses.
“We show that an interplay between two proteins, complexin and synaptotagmin, controls the vesicle fusion machinery in neurons, and that both proteins are necessary to trigger normal information flow and prevent uncontrolled spontaneous release,” says J. Troy Littleton, who led both studies and is an investigator in the Picower Institute for Learning and Memory and the Department of Biology and Department of Brain and Cognitive Sciences (BCS). The papers appear in the Dec. 2, 2012, and Jan. 2, 2013, issues of the Journal of Neuroscience.
Neurons have specialized needs, and one is to release neurotransmitters when the cell receives an electrical impulse that shoots down the axon to the synapses — typically in response to some stimulation. This impulse causes calcium to rush into the cell, which triggers the release of neurotransmitters across the synaptic gap to communicate with the next neuron. This neurotransmitter release is called an evoked response, as opposed to a spontaneous release (or “mini”), in which a small number of vesicles occasionally fuse without stimulation.
“So the first modification a neuron must make to the fusion machinery is to sense calcium,” says Jihye Lee, a postdoctoral associate in the Littleton lab and first author of the Jan. 2 paper that examines the role synaptotagmin plays in calcium sensing.
Synaptotagmin is a protein localized to the neuronal vesicles, with two calcium-binding domains, C2A and C2B. Lee examined how each domain functions in this role. C2B drives the fast fusion of vesicles with the membrane, and requires C2A to dive into the membrane and activate the fusion machinery that promotes mixing of the two lipid membranes.
The second major requirement for neurons is to prevent these fusion events until a calcium signal arrives. Otherwise, neuronal signals flood the brain and wreak havoc, which leads to such neurological disorders as epilepsy. “We found that a protein known as complexin binds to the fusion machinery and prevents it from working until the calcium signal comes,” says MIT affiliate Ramon Jorquera, first author of the Dec. 2 paper, which examines the interplay of complexin and synaptotagmin.
Complexin functions as a fusion clamp, keeping the vesicle from fusing with the synaptic membrane until synaptotagmin senses the influx of calcium and sets the extremely quick fusion process in motion.
This finding is important, Littleton says, because complexin is severely reduced in many neurological and psychological diseases, indicating these disease states may experience too many uncontrolled spontaneous release events. This reduction itself doesn’t cause the diseases, but it may contribute to the phenotypes.
The researchers conducted these studies using the fruit fly, a valuable model organism because of the ease of doing genetic manipulations and neuronal recordings. They created flies in which they deleted or over-expressed various combinations of the genes for the complexin and synaptotagmin proteins, which determined the contribution of each to evoked and spontaneous neurotransmitter release. For example, deleting the complexin clamp caused a 100-fold increase in spontaneous minis; taking away the calcium-sensing synaptotagmin protein eliminated it all.
The researchers also focused on a type of synapse that is representative of the majority of synapses in the human central nervous system — those that release the excitatory neurotransmitter glutamate.
“Because this same machinery appears to play a similar role in mammals, we think we can gain valuable understanding about how it is controlled in humans too,” Littleton says. “Our long-term goal is to learn how neurons normally talk to each other, and how this process goes awry during neurological and psychiatric diseases. This insight might ultimately allow us to restore proper synaptic function and brain communication in disease states.”
Sarah Huntwork-Rodriguez, Yulia Akbergenova and Richard W. Cho, all of the Picower Institute, BCS and the Department of Biology, also contributed to the Dec. 2 paper. Their work was supported by a National Institutes of Health grant and the PEW Latin American Fellows Program in the Biomedical Sciences.
Akbergenova and Zhuo Guan, of the Picower Institute, BCS and the Department of Biology, also contributed to the Jan. 2 paper. This work was supported by an NIH grant.
Two new MIT studies now demonstrate how neurons have adapted the cell’s standard fusion machinery to regulate the release of neurotransmitters at the neuron’s chemical junctions called synapses.
“We show that an interplay between two proteins, complexin and synaptotagmin, controls the vesicle fusion machinery in neurons, and that both proteins are necessary to trigger normal information flow and prevent uncontrolled spontaneous release,” says J. Troy Littleton, who led both studies and is an investigator in the Picower Institute for Learning and Memory and the Department of Biology and Department of Brain and Cognitive Sciences (BCS). The papers appear in the Dec. 2, 2012, and Jan. 2, 2013, issues of the Journal of Neuroscience.
Neurons have specialized needs, and one is to release neurotransmitters when the cell receives an electrical impulse that shoots down the axon to the synapses — typically in response to some stimulation. This impulse causes calcium to rush into the cell, which triggers the release of neurotransmitters across the synaptic gap to communicate with the next neuron. This neurotransmitter release is called an evoked response, as opposed to a spontaneous release (or “mini”), in which a small number of vesicles occasionally fuse without stimulation.
“So the first modification a neuron must make to the fusion machinery is to sense calcium,” says Jihye Lee, a postdoctoral associate in the Littleton lab and first author of the Jan. 2 paper that examines the role synaptotagmin plays in calcium sensing.
Synaptotagmin is a protein localized to the neuronal vesicles, with two calcium-binding domains, C2A and C2B. Lee examined how each domain functions in this role. C2B drives the fast fusion of vesicles with the membrane, and requires C2A to dive into the membrane and activate the fusion machinery that promotes mixing of the two lipid membranes.
The second major requirement for neurons is to prevent these fusion events until a calcium signal arrives. Otherwise, neuronal signals flood the brain and wreak havoc, which leads to such neurological disorders as epilepsy. “We found that a protein known as complexin binds to the fusion machinery and prevents it from working until the calcium signal comes,” says MIT affiliate Ramon Jorquera, first author of the Dec. 2 paper, which examines the interplay of complexin and synaptotagmin.
Complexin functions as a fusion clamp, keeping the vesicle from fusing with the synaptic membrane until synaptotagmin senses the influx of calcium and sets the extremely quick fusion process in motion.
This finding is important, Littleton says, because complexin is severely reduced in many neurological and psychological diseases, indicating these disease states may experience too many uncontrolled spontaneous release events. This reduction itself doesn’t cause the diseases, but it may contribute to the phenotypes.
The researchers conducted these studies using the fruit fly, a valuable model organism because of the ease of doing genetic manipulations and neuronal recordings. They created flies in which they deleted or over-expressed various combinations of the genes for the complexin and synaptotagmin proteins, which determined the contribution of each to evoked and spontaneous neurotransmitter release. For example, deleting the complexin clamp caused a 100-fold increase in spontaneous minis; taking away the calcium-sensing synaptotagmin protein eliminated it all.
The researchers also focused on a type of synapse that is representative of the majority of synapses in the human central nervous system — those that release the excitatory neurotransmitter glutamate.
“Because this same machinery appears to play a similar role in mammals, we think we can gain valuable understanding about how it is controlled in humans too,” Littleton says. “Our long-term goal is to learn how neurons normally talk to each other, and how this process goes awry during neurological and psychiatric diseases. This insight might ultimately allow us to restore proper synaptic function and brain communication in disease states.”
Sarah Huntwork-Rodriguez, Yulia Akbergenova and Richard W. Cho, all of the Picower Institute, BCS and the Department of Biology, also contributed to the Dec. 2 paper. Their work was supported by a National Institutes of Health grant and the PEW Latin American Fellows Program in the Biomedical Sciences.
Akbergenova and Zhuo Guan, of the Picower Institute, BCS and the Department of Biology, also contributed to the Jan. 2 paper. This work was supported by an NIH grant.