
A new study by researchers at the Picower Institute for Learning and Memory at MIT finds that the protein CPG2 is significantly less abundant in the brains of people with bipolar disorder (BD) and shows how specific mutations in the SYNE1 gene that encodes the protein undermine its expression and its function in neurons.
Led by Elly Nedivi, professor in MIT’s departments of Biology and Brain and Cognitive Sciences, and former postdoc Mette Rathje, the study goes beyond merely reporting associations between genetic variations and psychiatric disease. Instead, the team’s analysis and experiments show how a set of genetic differences in patients with bipolar disorder can lead to specific physiological dysfunction for neural circuit connections, or synapses, in the brain.
The mechanistic detail and specificity of the findings provide new and potentially important information for developing novel treatment strategies and for improving diagnostics, Nedivi says.
“It’s a rare situation where people have been able to link mutations genetically associated with increased risk of a mental health disorder to the underlying cellular dysfunction,” says Nedivi, senior author of the study online in Molecular Psychiatry. “For bipolar disorder this might be the one and only.”
The researchers are not suggesting that the CPG2-related variations in SYNE1 are “the cause” of bipolar disorder, but rather that they likely contribute significantly to susceptibility to the disease. Notably, they found that sometimes combinations of the variants, rather than single genetic differences, were required for significant dysfunction to become apparent in laboratory models.
“Our data fit a genetic architecture of BD, likely involving clusters of both regulatory and protein-coding variants, whose combined contribution to phenotype is an important piece of a puzzle containing other risk and protective factors influencing BD susceptibility,” the authors wrote.
CPG2 in the bipolar brain
During years of fundamental studies of synapses, Nedivi discovered CPG2, a protein expressed in response to neural activity, that helps regulate the number of receptors for the neurotransmitter glutamate at excitatory synapses. Regulation of glutamate receptor numbers is a key mechanism for modulating the strength of connections in brain circuits. When genetic studies identified SYNE1 as a risk gene specific to bipolar disorder, Nedivi’s team recognized the opportunity to shed light into the cellular mechanisms of this devastating neuropsychiatric disorder typified by recurring episodes of mania and depression.
For the new study, Rathje led the charge to investigate how CPG2 may be different in people with the disease. To do that, she collected samples of postmortem brain tissue from six brain banks. The samples included tissue from people who had been diagnosed with bipolar disorder, people who had neuropsychiatric disorders with comorbid symptoms such as depression or schizophrenia, and people who did not have any of those illnesses. Only in samples from people with bipolar disorder was CPG2 significantly lower. Other key synaptic proteins were not uniquely lower in bipolar patients.
“Our findings show a specific correlation between low CPG2 levels and incidence of BD that is not shared with schizophrenia or major depression patients,” the authors wrote.
From there they used deep-sequencing techniques on the same brain samples to look for genetic variations in the SYNE1 regions of BD patients with reduced CPG2 levels. They specifically looked at ones located in regions of the gene that could regulate expression of CPG2 and therefore its abundance.
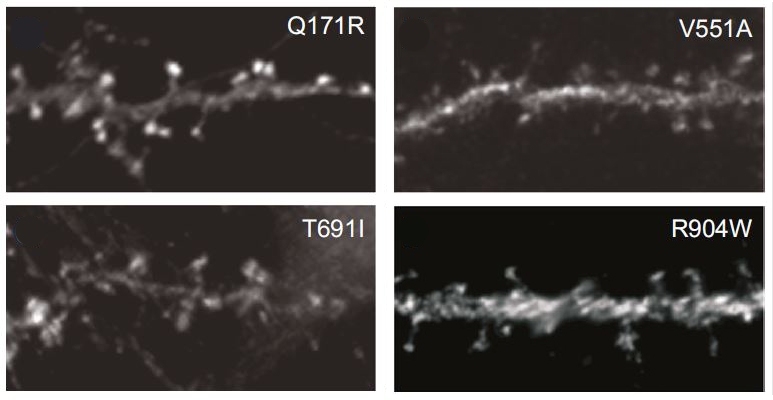
Meanwhile, they also combed through genomic databases to identify genetic variants in regions of the gene that code CPG2. Those mutations could adversely affect how the protein is built and functions.
Examining effects
The researchers then conducted a series of experiments to test the physiological consequences of both the regulatory and protein coding variants found in BD patients.
To test effects of non-coding variants on CPG2 expression, they cloned the CPG2 promoter regions from the human SYNE1 gene and attached them to a "reporter" that would measure how effective they were in directing protein expression in cultured neurons. They then compared these to the same regions cloned from BD patients that contained specific variants individually or in combination. Some did not affect the neurons’ ability to express CPG2 but some did profoundly. In two cases, pairs of variants (but neither of them individually), also reduced CPG2 expression.
Previously Nedivi’s lab showed that human CPG2 can be used to replace rat CPG2 in culture neurons, and that it works the same way to regulate glutamate receptor levels. Using this assay they tested which of the coding variants might cause problems with CPG2’s cellular function. They found specific culprits that either reduced the ability of CPG2 to locate in the “spines” that house excitatory synapses or that decreased the proper cycling of glutamate receptors within synapses.
The findings show how genetic variations associated with BD disrupt the levels and function of a protein crucial to synaptic activity and therefore the health of neural connections. It remains to be shown how these cellular deficits manifest as biopolar disorder.
Nedivi’s lab plans further studies including assessing behavioral implications of difference-making variants in lab animals. Another is to take a deeper look at how variants affect glutamate receptor cycling and whether there are ways to fix it. Finally, she said, she wants to continue investigating human samples to gain a more comprehensive view of how specific combinations of CPG2-affecting variants relate to disease risk and manifestation.
In addition to Rathje and Nedivi, the paper’s other authors are Hannah Waxman, Marc Benoit, Prasad Tammineni, Costin Leu, and Sven Loebrich.
The JPB Foundation, the Gail Steel Fund, the Carlsberg Foundation, the Lundbeck Foundation and the Danish Council for Independent Research funded the study.