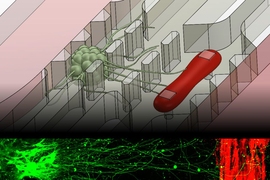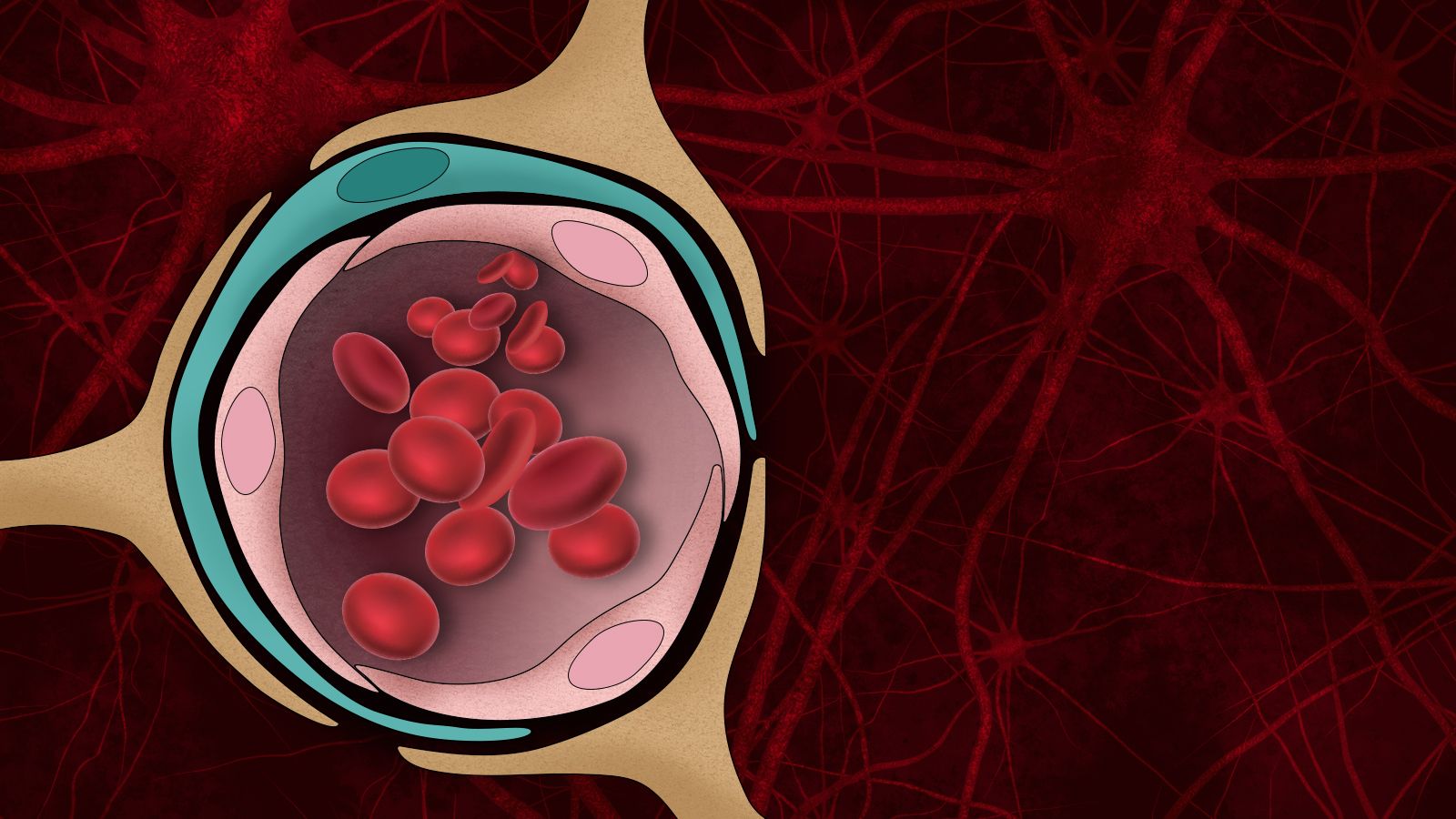

Beta-amyloid plaques, the protein aggregates that form in the brains of Alzheimer’s patients, disrupt many brain functions and can kill neurons. They can also damage the blood-brain barrier — the normally tight border that prevents harmful molecules in the bloodstream from entering the brain.
MIT engineers have now developed a tissue model that mimics beta-amyloid’s effects on the blood-brain barrier, and used it to show that this damage can lead molecules such as thrombin, a clotting factor normally found in the bloodstream, to enter the brain and cause additional damage to Alzheimer’s neurons.
“We were able to show clearly in this model that the amyloid-beta secreted by Alzheimer’s disease cells can actually impair barrier function, and once that is impaired, factors are secreted into the brain tissue that can have adverse effects on neuron health,” says Roger Kamm, the Cecil and Ida Green Distinguished Professor of Mechanical and Biological Engineering at MIT.
The researchers also used the tissue model to show that a drug that restores the blood-brain barrier can slow down the cell death seen in Alzheimer’s neurons.
Kamm and Rudolph Tanzi, a professor of neurology at Harvard Medical School and Massachusetts General Hospital, are the senior authors of the study, which appears in the August 12 issue of the journal Advanced Science. MIT postdoc Yoojin Shin is the paper’s lead author.
Barrier breakdown
The blood vessel cells that make up the blood-brain barrier have many specialized proteins that help them to form tight junctions — cellular structures that act as a strong seal between cells.
Alzheimer’s patients often experience damage to brain blood vessels caused by beta-amyloid proteins, an effect known as cerebral amyloid angiopathy (CAA). It is believed that this damage allows harmful molecules to get into the brain more easily. Kamm decided to study this phenomenon, and its role in Alzheimer’s, by modeling brain and blood vessel tissue on a microfluidic chip.
“What we were trying to do from the start was generate a model that we could use to understand the interactions between Alzheimer’s disease neurons and the brain vasculature,” Kamm says. “Given the fact that there’s been so little success in developing therapeutics that are effective against Alzheimer’s, there has been increased attention paid to CAA over the last couple of years.”
His lab began working on this project several years ago, along with researchers at MGH who had engineered neurons to produce large amounts of beta-amyloid proteins, just like the brain cells of Alzheimer’s patients.
Led by Shin, the researchers devised a way to grow these cells in a microfluidic channel, where they produce and secrete beta-amyloid protein. On the same chip, in a parallel channel, the researchers grew brain endothelial cells, which are the cells that form the blood-brain barrier. An empty channel separated the two channels while each tissue type developed.
After 10 days of cell growth, the researchers added collagen to the central channel separating the two tissue types, which allowed molecules to diffuse from one channel to the other. They found that within three to six days, beta-amyloid proteins secreted by the neurons began to accumulate in the endothelial tissue, which led the cells to become leakier. These cells also showed a decline in proteins that form tight junctions, and an increase in enzymes that break down the extracellular matrix that normally surrounds and supports blood vessels.
As a result of this breakdown in the blood-brain barrier, thrombin was able to pass from blood flowing through the leaky vessels into the Alzheimer’s neurons. Excessive levels of thrombin can harm neurons and lead to cell death.
“We were able to demonstrate this bidirectional signaling between cell types and really solidify things that had been seen previously in animal experiments, but reproduce them in a model system that we can control with much more detail and better fidelity,” Kamm says.
Plugging the leaks
The researchers then decided to test two drugs that have previously been shown to solidify the blood-brain barrier in simpler models of endothelial tissue. Both of these drugs are FDA-approved to treat other conditions. The researchers found that one of these drugs, etodolac, worked very well, while the other, beclomethasone, had little effect on leakiness in their tissue model.
In tissue treated with etodolac, the blood-brain barrier became tighter, and neurons’ survival rates improved. The MIT and MGH team is now working with a drug discovery consortium to look for other drugs that might be able to restore the blood-brain barrier in Alzheimer’s patients.
“We’re starting to use this platform to screen for drugs that have come out of very simple single cell screens that we now need to validate in a more complex system,” Kamm says. “This approach could offer a new potential form of Alzheimer’s treatment, especially given the fact that so few treatments have been demonstrated to be effective.”
The research was funded by the Cure Alzheimer’s Fund and the JPB Foundation.