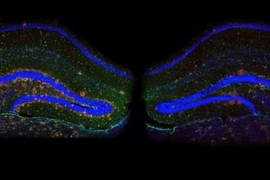
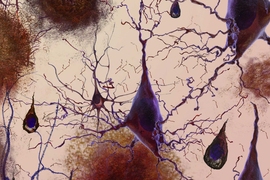
Our susceptibility to disease depends both on the genes that we inherit from our parents and on our lifetime experiences. These two components — nature and nurture — seem to affect very different processes in the context of Alzheimer's disease, according to a new study published today in the journal Nature.
The study was carried out by an interdisciplinary team at MIT and the Broad Institute, and was co-led by Li-Huei Tsai, the Picower Professor at MIT and director of the Picower Institute for Learning and Memory, and Manolis Kellis, a professor in MIT's Computer Science and Artificial Intelligence Laboratory (CSAIL).
The researchers analyzed changes that occur in genes and in regions that regulate genes as Alzheimer’s disease progresses, using a mouse model of Alzheimer’s disease that Tsai’s lab originally developed several years ago. The mice were engineered so that the gene for a protein called p25 can be overstimulated in the brain, which prompts the mice to develop symptoms very similar to Alzheimer’s disease in humans.
“These programmable mice allowed us to study, for the first time, the changes occurring during early stages of the disease, before symptoms even begin to appear,” Tsai says. “We could then compare them to changes in later stages of the disease, when neurodegeneration and cognitive impairment are evident.”
Opposing changes
The researchers profiled multiple chemical modifications, known as epigenetic marks, in the hippocampus of mice expressing too much p25 and compared them with control mice. These epigenetic marks reveal the activity of diverse genomic regions — in particular, the regulatory control regions that control the expression of nearby genes. The researchers also directly profiled the levels of all genes.
“We found two opposing signatures associated with disease progression that are consistent with the pathophysiology of Alzheimer’s disease,” says Elizabeta Gjoneska, joint first author of the paper and a postdoc at the Picower Institute. “Neuronal plasticity processes that are involved in learning and memory were dampened, and immune and inflammatory pathways were activated.”
The active regions specifically matched regions active in a type of immune cells known as microglia, which are responsible for clearing away infected or damaged cells. They also secrete chemicals that produce inflammation.
“Our data suggest that microglia are heavily activated during Alzheimer’s disease progression, although it is unknown exactly how they contribute to the disease,” Tsai says. “These cells are important for normal brain function and share their key cell-surface markers, CD14, with macrophages that infiltrate the brain from elsewhere in the body during disease progression.”
Conserved epigenomic signatures
The researchers then compared the results in mice with what is known about Alzheimer’s disease in humans. They found that differences in gene levels in the Alzheimer’s-like mouse brain matched differences previously seen in the brains of Alzheimer's patients, which prompted them to ask if the epigenetic signatures might also be conserved.
The researchers found that this was the case — specifically, the same regulatory regions that were active or repressed in mice showed the same patterns in humans. They also found that the regions with increased activity in the mouse model of Alzheimer’s disease had immune functions in humans, and the regions that showed decreased activity had neural functions in humans.
“Our results show that functional conservation between human and mouse is not restricted to protein-coding genes,” says Andreas Pfenning, joint first author of the study and a postdoc at MIT. “This opens up the use of epigenomics methods in model organisms to study an inaccessible organ like the brain, and how it changes in response to activity or disease.”
Genetic variants cluster in immune pathways
Previous studies of the genomes of Alzheimer’s patients had identified common genetic variants associated with the disease, but scientists did not know how these DNA variants could contribute to the disease, since the majority of them are found outside of protein-coding regions.
“Our conserved epigenomic maps allowed us to now place these noncoding genetic variants in the context of disease-relevant regulatory regions and interpret their contribution to the disease predisposition,” Kellis says. “As inherited common genetic variants always precede disease onset, they are always indicative of causal roles, and thus can shed additional light on the epigenomic alterations.”
The researchers found that genetic variants associated with Alzheimer's disease were only associated with immune processes, and not with neural processes, indicating that genetic predisposition to Alzheimer's disease primarily affects the circuitry of immune processes, rather than neuronal processes.
“Our results suggest that repression of neural pathways does not represent genetic predisposition, even though it is a hallmark of Alzheimer's,” Tsai says. “Instead, it may occur as a consequence of environmental factors and aging, and result from interactions with the altered immune pathways.”
The researchers identified a small number of master regulators that target many of the regulatory regions that overlap Alzheimer's-associated genetic variants in humans. Among these, PU.1 targets a large number of altered regulatory regions, and the genetic region encoding PU.1 is associated with Alzheimer's disease, suggesting PU.1 as a potential therapeutic target.
“The new focus on immune-cell types, and the specific regulators uncovered, provide new therapeutic avenues,” Kellis says. “Moreover, the conservation of epigenomic signatures between mouse and human provides a platform upon which we can test such therapeutics and their effect on cognition, pathology, and the epigenomic signatures of Alzheimer's.”
“In an elegant series of experiments, the paper adds to a growing body of evidence, which began with genomewide association studies, implicating immune function in the pathogenesis of Alzheimer’s disease. This potentially paves the way for exploiting peripheral myeloid cells to develop biomarkers and therapeutics to target the disease,” says David Bennett, a professor of neurological sciences at Rush University Medical Center, who was not part of the research team.
The research was a component of the National Institutes of Health’s Roadmap Epigenomics Program and was also funded by the Belfer Neurodegeneration Consortium and the Swiss National Science Foundation. The paper was published along with another, whose senior author was Kellis, reporting on the integrative analysis of 111 reference epigenomes. It joins a total of 24 related papers published this week in the Nature and other Nature journals.
Other authors are CSAIL postdoc Gerald Quon, Picower postdoc Hansruedi Mathys, and former CSAIL research scientist Anshul Kundaje.